Update from Day 1 of the Nanopore Community Meeting in New York: infectious disease, first human genomes on a MinION, Clive Brown plenary, more
Thursday was a busy day at the Nanopore Community meeting in New York. 200 attendees travelled to the Metropolitan Pavilion to listen to plenaries, take part in breakout sessions and view the technology showcase.
The day started with two plenary talks on infectious disease.
Professor Jane Carlton from New York University described how the MinION fits into her work with malaria and how she has used the MinION to sequence Plasmodium in the field. Her goal is to provide a method for real-time field-based malaria parasite detection. Her group developed a technique for preparing and sequencing malaria DNA at New York University using blood samples, and then used the technique in the field in India. They were able to sequence malaria parasites within three hours of a patient being diagnosed. Their on-going work includes identifying patients infected with multiple species of malaria, identifying SNPs associated with drug resistance and analysing human genes related to parasite infection (eg DARC).

Our second plenary speaker, Dr Tariq Sadiq, Reader and Consultant Physician at St George’s, University of London discussed the challenges of controlling Sexually Transmitted Infections (STIs) with a focus on gonorrhoea, where there is inadequate access to prompt diagnosis and treatment for both patients and their partners. Poor surveillance of STIs in communities around the world restricts public health interventions and the effectiveness of strategies to fight emerging antimicrobial resistance (AMR). Gonorrhoea can lead to blindness and infertility, patients need to begin treatment as soon as possible, determining drug resistance (as well as susceptibility) of their infection allows for responsible stewardship of available therapies – including the use of antibiotics that are currently overlooked due to prevalent resistance in a community. Dr Sadiq described a project at St George’s to evaluate the MinION as a tool for AMR prediction and phylogeny reconstruction for real-time surveillance of outbreaks. The MinION’s portable, real-time data combined with the ease and speed of the 1D rapid kit has the potential to optimize treatment strategies, which may ultimately give scope for reducing STI transmission and the burden of treatable disease world-wide.
The first breakout session on whole genome sequencing and metagenomics featured speakers across the spectrum of field-based environmental analyses to central laboratories.
Dr. Arwyn Edwards of Aberystwyth University presented his research on glacial microbial communities and impressed the importance of fast, portable, on-site analysis of these rapidly changing landscapes. Microbial communities contribute to the behaviour of glaciers, whether through accelerating recession or dramatic colour change but are not well understood. Dr. Edwards and his team are establishing a baseline so that these changes can be monitored over time. His group travelled to the High Arctic (78 degrees north) to gather samples and perform MinION sequencing and species classification in situ in these extreme environments along with their ‘Metagenomad’ backpack and polar bear deterrant (bringing new meaning to shotgun metagenomics). The results from their mobile laboratory compared favourably with traditional lab based technologies and the group continues to optimise wet protocols and bioinformatics approaches for ‘off-grid’ metagenomic sequencing, raising the prospect of characterizing Earth’s microbiomes at source. A genuine example of “sequence everything, sequence everywhere”.

Alex Dilthey from NHGRI presented mashmap - a fast, alignment-free approximate mapping algorithm for long reads. Mashmap builds a sparse index of a basecalled read (using 5-10% of kmers appearing in that read) and then uses the sparse index to identify all regions of a reference to which the read maps. This tool has several features including a constant-time mapping setting where the number of k-mers in the index is fixed independent of read length; a mapping quality determination using the Bayesian probability of each returned mapping position and a quality-aware phylogenetic assignment.
Vladimir Benes of the German European Molecular Biology Laboratory (EMBL), spoke about the continued challenges of complete de novo assembly of ‘larger’ genomes and the impact of this to an extended community around a core lab. Dr Benes described recent work at EMBL using nanopore sequencing to finish several de novo genome sequencing projects. By designing protocols to support >100kb reads, the assembly of several 0.5-1Gb genomes has been improved. Indications from first use of the new R9 are that the group will be able to reduce the amount of data generated by short-read sequencing technology needed to generate worthwhile genome assemblies.
For example, they assembled a BAC clone of microsatelite-rich Hstx1/Hstx2 region of the mouse X chromosome with just 76 reads. Insect cell lines used for expression of eukaryotic recombinant proteins, lack of genomic characterisation limits the amount of tinkering possible. Used a multi-omic approach to charaterise 2 cell lines, addition of nanopore data improved the assembly significantly.
Peter Thielen presented on optimised metagenomics methods for influenza genomic surveillance from rapid sample prep to analysis. Influenza is a major human pathogen killing 20,
Alex Salazar from TU Delft reviewed the utility of long reads to better understand the genomes of organisms with higher chromosome ploidies. He described the ability to achieve more contiguous assemblies and to provide phasing information that may be critical for deciphering genetic differences across haplotypes, i.e. different copies of a chromosome. Traditional short-read technologies tend to yield fragmented assemblies and are harder to reconstruct haplotypes from. The Delft group has used the MinION to reconstruct the genome architecture of S. pastorianus — an industrial yeast species with two sub-genomes, varying chromosome copy number, rearrangements and fusions. MinION sequencing alone enabled them to efficiently obtain telomere-to-telomere consensus reconstructions of chromosomes in this organism.
Using the data from a single flow cell he achieved an almost complete assembly of a haploid S. cervisae genome. He then moved on to the more challenging genome of S. pastorianus. A hybrid aneuploid genome, it's the result of the fusion of 2 genomes that were about 85% identical and subsequent change in chromosome cop-number. Using a combination of library preps he was able to get almost complete assembly and could identify multi-gene deletions by looking at assembly graph.
Reviewing recent advancements in MinION chemistry, he reflected that individual sequencing runs are often sufficient to accurately infer copy number variation.
Minh Duc Cao from the University of Queensland described the utility of long reads that span most repeat sequences and therefore help complete fragmented short read assemblies. npScarf allows users to terminate data acquisition at the point the data is sufficient for an assembly of a specified quality and completeness. This tool is available at: https://github.com/mdcao/npScarf

Miten Jain is one of the authors on the recent comprehensive review of nanopore sequencing in Genome Biology. Today he discussed improvements to MinION sequencing performance since 2014. Miten also spoke about detection of native base modifications C vs 5mC vs 5hmC and A vs m6A. Their research group has shown that MinION data can detect local and global changes in the proportion of methylation in e.coli that occur during different growth phases. Miten presented some work in training a mismatch matrix for aligning reads generated on the MinION using the new direct RNA sequencing kit just released to a few developers.Continuing the theme of epigenetic analysis,
Winston Timp from Johns Hopkins University described mechanisms by which nanopore sequencers can directly identify base modifications, without the need for the creation of cDNA. He discussed his group’s work to use a hidden Markov model to distinguish 5-methylcytosine from unmethylated cytosine in DNA. This method has allowed them to measure global patterns of methylation from low-coverage sequencing and quantify the methylation status of CpG islands from single MinION reads. He presented an analysis of methylation patterns over very long reads from a reduced-representation tumor/normal pair.
He showed results on an MCF10A breast cancer cell line that revealed some strands are hypermethylated but others have lower levels of methylation. He showed that R9 chemistry is improving the accuracy of methylation detection over R7.
Over lunch, the NCM attendees spent time in the technology showcase area, meeting the MinION, PromethION, VolTRAX and analysis tools including npReader and MinoTour.
Dan Turner, Oxford Nanopore’s Senior Director of Applications, focused on two emerging themes for nanopore: Direct RNA sequencing and VolTRAX, the programmable device for automated sample and library preparation. He reminded the audience that there are now a wide range of library prep options available – these address any experiment that researchers would wish to do with nanopore technology. For community members, there is a selector tool that helps you decide which one is most suited to your experiment. This includes rapid library prep, from DNA to library in 5-10 minutes, and coupled with rapid DNA extraction it’s possible to go from cells to ID in 20 minutes. Dan also described the new 16S analysis workflow that has just been released. Reviewing the unique capability of nanopores to perform direct RNA sequencing, Dan outlined the ambition to achieve a million direct RNA reads on a MinION run – each of which is a full transcript.
Finally, Dan reviewed VolTRAX – the automated library and sample prep device that is now being shipped to early users. VolTRAX will first be used for 1D and 2D library prep, but the direct RNA prep will be released shortly after. He showed that DNA extraction and PCR can be performed on Voltrax, moving towards the goal of enabling anyone to sequence anything, anywhere.
Birds of a Feather! A new format of informal discussion, where the topics are decided by the attendees of the meeting.

After coffee, we moved to three more plenaries – for the first time speakers were presenting human genomes that had been sequenced on the MinION. Clive Brown, Oxford Nanopore’s CTO, gave his plenary talk, which you can now watch below.
Human genomes on a MinION
In the afternoon, the first human genomes to be sequenced on a MinION were presented. The presenters noted meaningful first results as well as a desire to continue work on expanding their projects and share their results with the scientific community. See their presentations/resources below:
Wigard Kloosterman
Center for Molecular Medicine, UMC Utrecht
Characterization of structural variations and chromothripsis in nanopore sequencing data of human genomes
Michael Simpson
Genomics Plc and the Wellcome Trust Centre for Human Genetics
Nanopore sequencing of human genomes
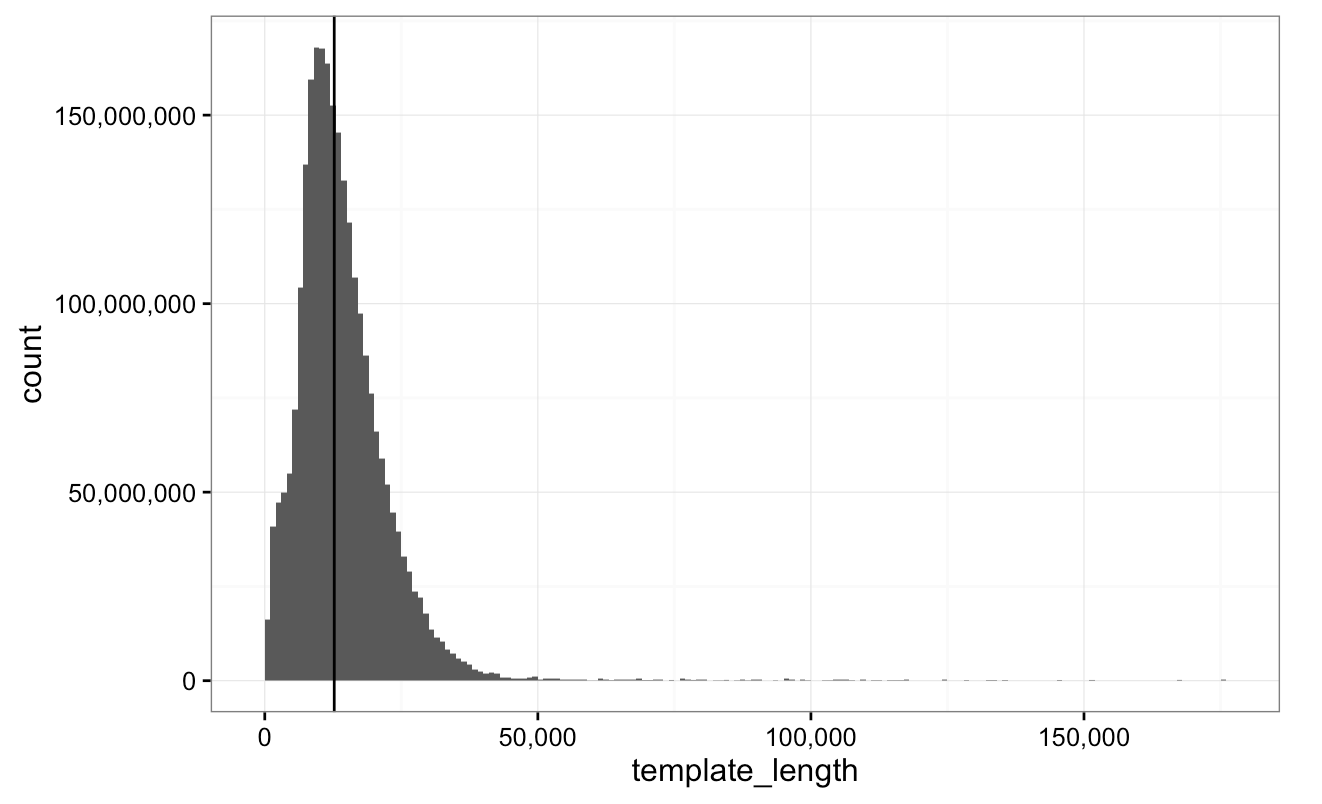
Nanopore-WGS-Consortium (8 centres)
NA12878 genome sequence (20X; gDNA) using the MinION
View on github| Over drinks, the audience heard from Rachelle Jensen of Indigo V expeditions on the impact of genomic analysis on ocean research. Having recently used the MinION the group are excited about performing research at sea, making expeditions much more efficient as they can direct the next stage of research in real time, rather than on subsequent expeditions if using non-portable technologies. Indigo V works with a network of sailing citizen scientists and outlined their vision for a MinION (or SmidgION) and VolTRAX aboard a network of boats, creating a real time ocean biology surveillance network. |






