Rapid surveillance of Orthopoxvirus monkeypox from complex metagenomic samples
- Published on: November 10 2022
International travel and urbanisation help novel pathogens enter new geographical areas. Researchers around the world are applying metagenomic analyses to identify and characterise emerging pathogens to better understand their epidemiology, transmission patterns, evolution, and adaptation to human transmission.
The first mpox sample sequenced with nanopore
Since May 2022, mpox — a zoonotic virus endemic to the African continent — caused by infection with Orthopoxvirus monkeypox (MPXV), has been reported in over 30 countries outside of Africa. At the University of São Paulo in Brazil, Ingra Morales Claro and her team used nanopore sequencing to study the first confirmed case of mpox in Brazil1. The team isolated viral DNA from a clinical research sample taken from skin lesions and prepared sequencing libraries using the Rapid PCR Barcoding Kit. Libraries were then sequenced on a MinION, producing a total of >950,000 reads and enabling a turnaround of 18 hours from DNA extraction to consensus sequence generation.
‘The average [read] depth was 277.7x, covering 100% of the viral genome with at least 1 read, and an N50 of 4,493’
Claro, I.M. et al. Rev. Inst. Med. Trop. Sao Paulo (2022)
The almost complete 197 kb genome assembly revealed that it clustered within clade III (the newly proposed B.1 lineage), which is associated with West Africa, and is closely related to MPXV sequences from Portugal, Germany, USA, and Spain. MPXV in clade III is associated with lower disease severity than those in clade I, which cluster in Central Africa.
Determining transmission patterns
Joana Isidro and her colleagues at the National Institute of Health in Portugal are also combining metagenomics with nanopore sequencing. The team generated the first draft genome sequence of an MPXV associated with the recent major outbreak in Portugal2. Using DNA isolated from skin lesions, libraries were prepared using the PCR-free Rapid Barcoding Sequencing Kit and sequenced on a MinION device. Sequencing reads were analysed and mapped to a reference genome in real time using RAMPART — delivering immediate visualisation of genome coverage. Approximately 800,000 reads were generated within 18 hours, of which about 0.5% were identified as MPXV.
A comparison with the reference sequence from the 2018–19 multi-country MPXV outbreak confirmed the presence of approximately 50 SNPs in the newly sequenced MPXV from Portugal. This substitution rate is far greater than expected for Orthopoxviruses, suggesting that ‘viral genome sequencing might provide sufficient resolution to track the transmission dynamics and outbreak spread, which seemed to be challenging for a presumably slow-evolving double-stranded DNA virus’3. Phylogenetic analyses suggested the new sequence was closely related to viruses previously imported from Nigeria to the UK, Israel, and Singapore in 2018–19, and belonged to clade III (Figure 1)2,3.
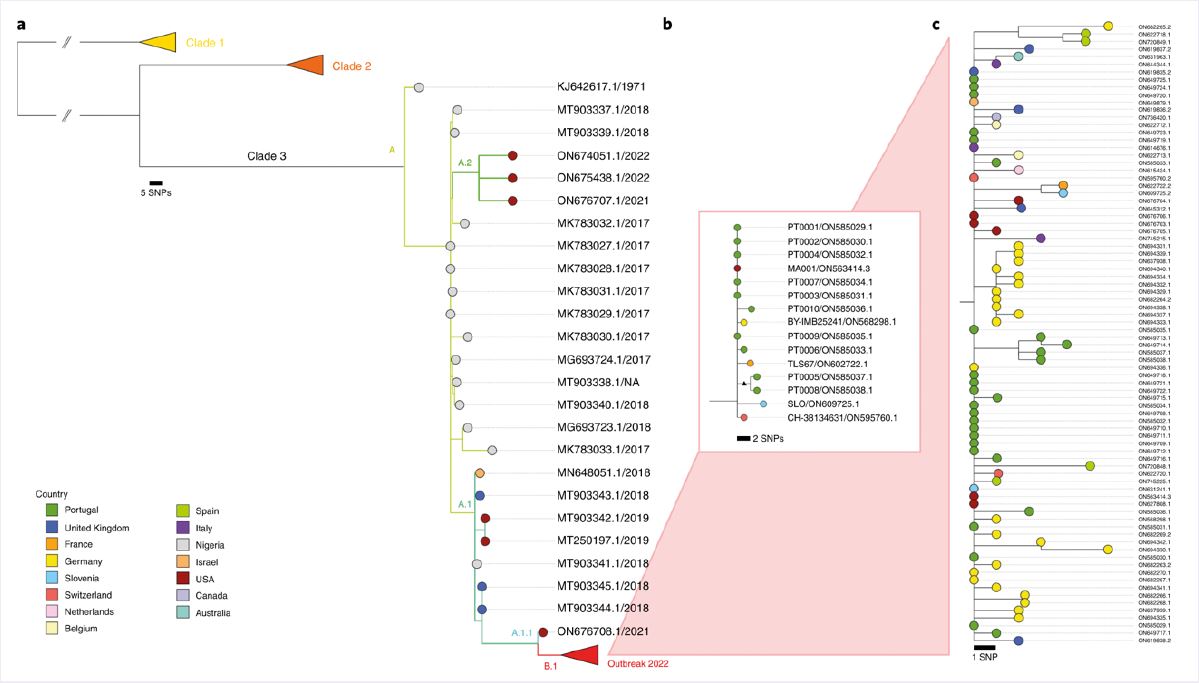
Figure 1: Phylogenetic analysis of MPXV viral sequences associated with the 2022 worldwide outbreak. a) Global phylogeny showing that the 2022 outbreak cluster (lineage B.1) belongs to clade 3. b) Genetic diversity within the outbreak cluster, including the first outbreak-related MPXV genome sequenced in Portugal and all available outbreak genome sequences released to the National Center for Biotechnology Information (NCBI) by 27 May 2022. c) Updated phylogenetic tree with sequences available in the NCBI as of 15 June 2022. Image taken from Isidro et al.3 and available under Creative Commons license (creativecommons.org/licenses/by/4.0).
‘Emergence of new viral infections with significant public health impact are frequent events, which re-enforces the need for comprehensive methodologies to detect rare, novel or emerging pathogens’
Alcolea-Medina, A. et al. SSRN (2022)
Rapid metagenomic sequencing
Using nanopore technology, Adela Alcolea-Medina and her team at the Centre for Clinical Infection & Diagnostics Research, UK, developed a simple metagenomic workflow to detect low-abundance RNA and DNA viruses in different sample types4. Their approach applied genome sequencing using long nanopore reads and required only seven hours from sample receipt to answer — offering a significant advantage over metagenomic detection using traditional short-read sequencing technologies, which typically take three to five days. The method also demonstrated the potential to differentiate between viruses that present with similar symptoms.
The streamlined workflow comprised library preparation, using the Rapid PCR Barcoding Kit, before sequencing on a GridION device, and live basecalling using MinKNOW for real-time analysis. Testing the workflow on four MPXV-positive nasopharyngeal research samples, the team detected MPXV in all four samples within 30 minutes of sequencing. To test their method using samples of unknown aetiology, the team analysed four research samples from blistering skin rashes: MPXV was detected in three samples within minutes of sequencing. In two of the samples, 94% and 96% of the viral genome were recovered at ≥10x depth after 16 hours of sequencing.

For the third research sample, only 49% of the genome was recovered, which may reflect the stage of disease at sample collection — day 18 of MPXV infection and receiving antiviral treatment. Varicella zoster virus (VZV), which causes chickenpox, was detected within 30 minutes of sequencing the fourth sample: 100% of the genome was sequenced to ≥10x depth of coverage.
These studies reveal how nanopore sequencing is supporting researchers to rapidly identify and fully characterise pathogens from complex metagenomic samples — providing actionable results to inform public health responses.
1. Claro, I.M. et al. Shotgun metagenomic sequencing of the first case of monkeypox virus in Brazil, 2022. Rev Inst Med Trop Sao Paulo. 24;64:e48 (2022).
2. Isidro, J. Monkeypox virus genome sequencing. Presentation. Available at: https://nanoporetech.com/resource-centre/monkeypox-virus-genome-sequencing-first-draft-genome-associated-2022-multi-country [Accessed 10 Oct 2022]
3. Isidro, J., et al. Phylogenomic characterization and signs of microevolution in the 2022 multi-country outbreak of monkeypox virus. Nat. Med. 28, 1569–1572 (2022).
4. Alcolea-Medina, A. et al. Novel, rapid metagenomic method to detect emerging viral pathogens applied to human monkeypox infections. doi.org/10.2139/ssrn.4132526 (2022).






