Metagenomic analysis of viral outbreaks case study
- Published on: March 22 2019
In recent decades, human disease outbreaks caused by arboviruses have increased in prevalence. Arboviruses are predominantly RNA viruses that replicate within blood-sucking arthropod vectors. Dengue virus (DENV) and chikungunya virus (CHIKV), transmitted to humans via Aedes species mosquitoes, are two arboviruses of increasing concern as they no longer require enzootic amplification and have been responsible for widespread epidemics1.
DENV and CHIKV are single-stranded positive-sense RNA viruses with overlapping geographical distributions. Differential identification based on symptoms is difficult as the clinical presentations are similar (e.g. high fever, rash, headache, myalgia)1. Therefore, rapid and unbiased identification methods are vital for determining the pathogen responsible for an outbreak, for real-time genomic surveillance and the ability to discriminate co-infection.
Metagenomic sequencing using nanopore technology has the power to detect and monitor syndromic outbreaks in a single unbiased and real-time assay. Kafetzopoulou and colleagues tested the feasibility and sensitivity of direct metagenomic sequencing of DENV and CHIKV genomes in serum and plasma samples infected with a range of viral loads1,2. Four samples for each virus were selected for nanopore sequencing, which provided a 99% genome coverage at a depth of 20x, even for samples with very low viral titres (DENV 31.29Ct; CHIKV 32.52Ct). High concordance was observed between short-read and nanopore sequencing data (Figure 1).
Importantly, using nanopore sequencing, near maximum coverage for both viruses was obtained within just 8 minutes when using the 1D2 Sequencing Kit or within 85 minutes when using the Rapid Sequencing Kit for library preparation. Furthermore, in one CHIKV sample the researchers identified the presence of DENV, highlighting the significant advantage that nanopore sequencing has over targeted techniques in its ability to detect co-infection. Taken together, these results demonstrate the sensitivity and speed that nanopore sequencing technology provides, and the researchers conclude that nanopore technology is:
‘… ideally suited for the investigation of viral species with high levels of genetic diversity which are difficult to assess using targeted methods'
Kafetzopoulou, L.E. et al. Euro. Surveill. (2018)
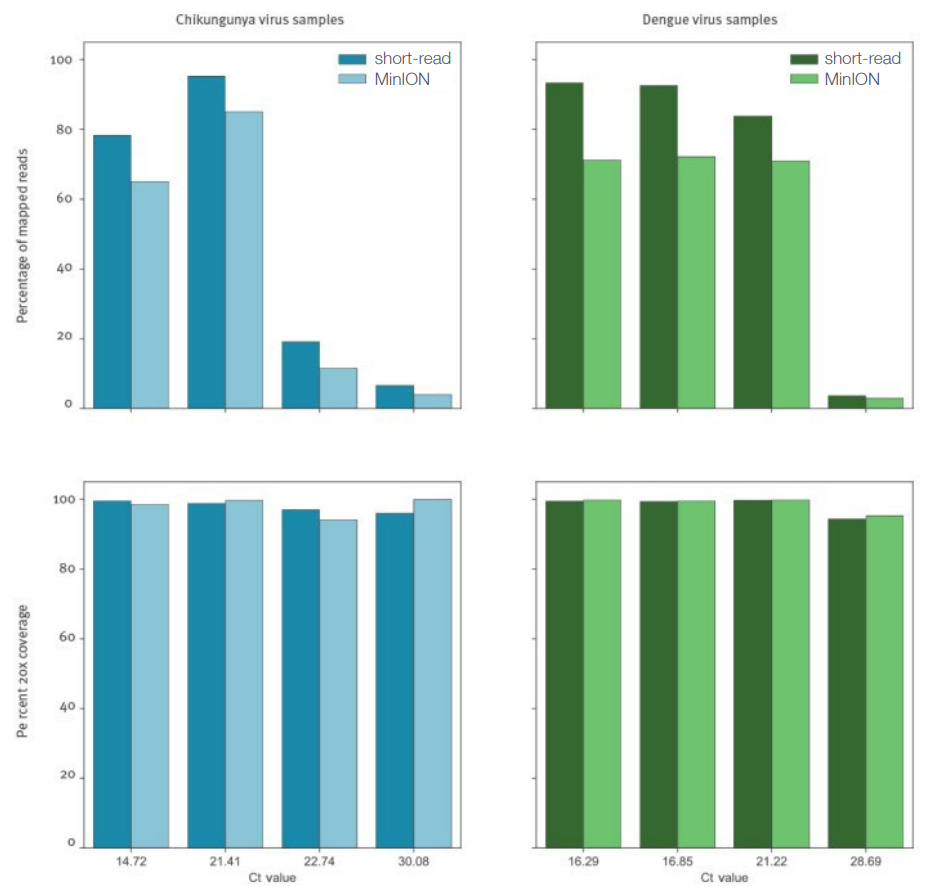
Figure 1: Comparison of the proportion of viral reads and reference genome coverage between nanopore and short-read sequencing data, demonstrating highly concordant results between the techniques and near 100% coverage at low viral titres. For each sample, identified by Ct value, the percentage of total reads mapping to the appropriate reference sequence, and therefore attributed as viral in origin, is plotted in the upper panels. Lower panels display the percentage of the reference genome sequenced to a minimum depth of 20-fold in the data generated. Adapted from Kafetzopoulou et al 1.
A significant advantage of nanopore sequencing using the portable MinION device is the facility for direct field application, which allows rapid deployment and negates the need to ship samples back to the laboratory. For example, in-field genomic surveillance of an ongoing Ebola epidemic was achieved by Quick et al. through successful application of nanopore sequencing technology3. Kafetzopoulou and colleagues travelled to the Institute of Lassa Fever Research and Control in Nigeria, a remote and resource- limited location, to apply their nanopore sequencing workflow to in-field monitoring of a Lassa viral outbreak4. Lassa fever is endemic in Nigeria and the virus is transmitted via contact with infected rats.
The Lassa virus genome is highly divergent and current detection requires two real-time, reverse transcription PCRs to cover all the possible variants of Lassa being circulated. Unbiased, rapid metagenomic analysis using nanopore technology is ideally suited for such situations, where investigation of highly genetically diverse viral species is cumbersome using targeted methods. During their time in Nigeria, the team encountered the largest Lassa fever outbreak ever reported. They established a nanopore sequencing and analysis workflow in a resource-limited setting, delivering complete genomic analysis of 35 samples within 21 days from the request of sequencing information on circulating strains by the Nigerian Centre of Disease Control. Phylogenetic analysis of the virus indicated that the outbreak was due to independent spill-over from the rodent reservoir, with no evidence of extensive human-to-human transmission. Kafetzopoulou and colleagues concluded that their workflow:
‘… confirms the feasibility of field metagenomic sequencing for these and likely other RNA viruses, highlighting the applicability of this approach to front-line public health'
Liana Kafetzopoulou, Public Health England, UK
1. Kafetzopoulou, L.E. et al. Assessment of Metagenomic MinION and Illumina sequencing as an approach for the recovery of whole genome sequences of chikungunya and dengue viruses directly from clinical samples. Euro. Surveill. 23(50) (2018).
2. Kafetzopoulou, L.E. et al. Metagenomic sequencing at the epicenter of the Nigeria 2018 Lassa fever outbreak. Science. 363(6422):74-77. (2019).
3. Quick, J. et al. Real-time, portable genome sequencing for Ebola surveillance. Nature 530(7589):228-232 (2016).
4. Kafetzopoulou, L.E. Metagenomic nanopore sequencing: RNA viruses from lab to field. Presentation. [Accessed: 30 October 2018]






