Mapping and phasing structural variation

- Published on: October 16 2018
Accounting for a far greater number of variable bases than single nucleotide variations (SNVs), structural variation (SV) is an important class of genetic variation that has been implicated in a wide range of genetic disorders. To address the limitations of short-read sequencing technology to accurately and cost-effectively characterise SV, an international research team led by Dr. Wigard Kloosterman of the University Medical Centre Utrecht, assessed the performance of long-read sequencing delivered by nanopore technology1 . The team performed whole genome sequencing of two DNA samples using both the MinION and a short-read sequencing technology. The samples were obtained from individuals with congenital disease resulting from complex chromothripsis, which is characterised by dozens of locally clustered genomic rearrangements affecting one or a few chromosome(s) in a cell.
In one sample, nanopore sequencing at 16x depth allowed the detection of all of the previously validated de novo chromothripsis breakpoint junctions. For the second sample, which was sequenced at 11x depth, the nanopore data allowed 24 of 29 previously validated breakpoints to be detected; however, further investigation revealed that two of the 5 undetected breakpoints represented a complex combination of joined segments which had been incorrectly assigned in the long-insert mate pair validated data set — further highlighting the benefits of long sequencing reads (Figure 1).
Detection of the remaining breakpoint junctions was hampered by insufficient depth of coverage. In total, in the second sample, nanopore sequencing at 11x depth allowed the detection of 29 of 31 (91%) breakpoint junctions, which compared favourably to the 22 (69%) detected using short-read sequencing at 30x coverage. Furthermore, four validated breakpoint junctions were only detected using nanopore sequencing and were not found in either the long-mate pair or shortread data set. By subsampling their data, the team were able to identify 14x depth of coverage as the minimum required to detect all breakpoint junctions using nanopore sequencing.
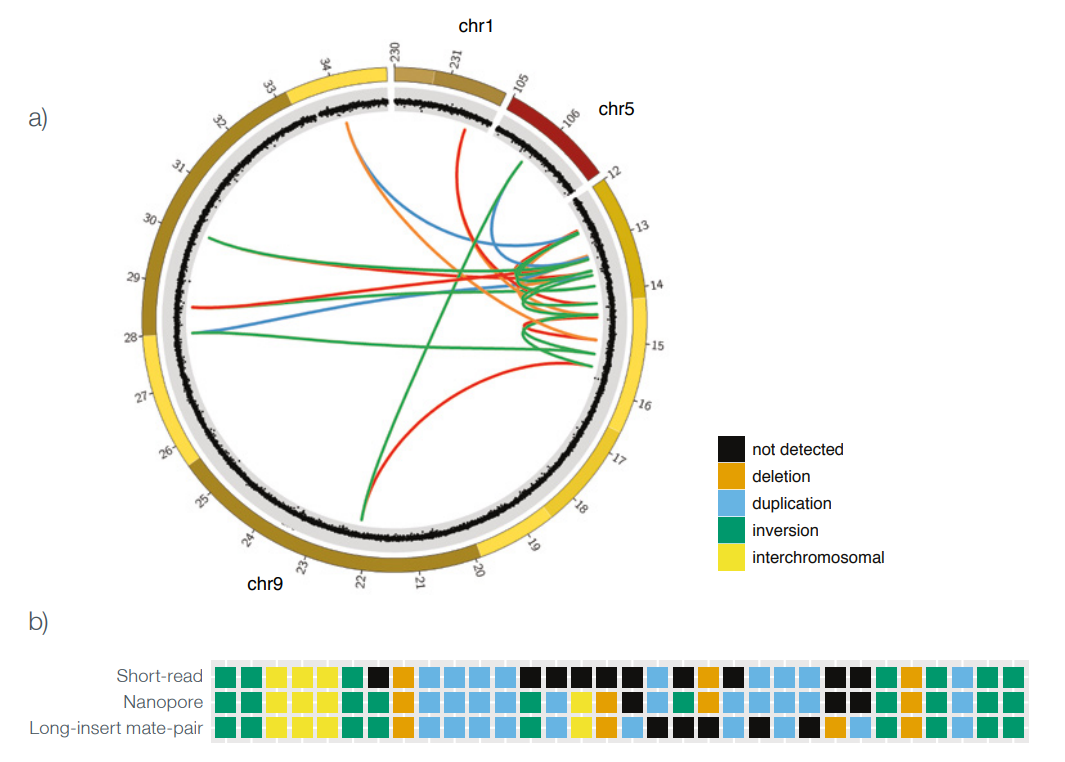 Figure 1: Nanopore sequencing accurately detects more chromothripsis breakpoints than alternative sequencing approaches. a) Circos plot of all breakpoint junctions in a complex chromothripsis sample. b) Comparison of different sequencing approaches to genotype breakpoint junctions. SVs were detected in short-read and nanopore data using the Delly and NanoSV tools respectively. Figure adapted from Cretu Stancu et al. 1
Figure 1: Nanopore sequencing accurately detects more chromothripsis breakpoints than alternative sequencing approaches. a) Circos plot of all breakpoint junctions in a complex chromothripsis sample. b) Comparison of different sequencing approaches to genotype breakpoint junctions. SVs were detected in short-read and nanopore data using the Delly and NanoSV tools respectively. Figure adapted from Cretu Stancu et al. 1Phasing of all chromothripsis breakpoints demonstrated paternal origin.
An important benefit of long nanopore sequencing reads is the facility for phasing. It had previously been hypothesised that germline chromothripsis originates from paternal chromosomes; however, this was based on only a few breakpoint junction sequences or deletions. Using nanopore sequencing, the team were able to phase all of the chromothripsis breakpoints detected, identifying their paternal origin and thereby providing further weight to the earlier hypothesis.
In the course of this research, the performance of a number of long-read SV calling tools was assessed, with the team demonstrating that their in-house developed tool, NanoSV, provided superior performance over a range of experimental parameters. Summarising their research, the team suggest that their work: ‘demonstrates the potential of long-read, portable sequencing technology for human genomics research and clinical applications’.*
* Nanopore devices are currently for research use only
This case study is taken from the human white paper.
1. Cretu Stancu, M. et al. Mapping and phasing of structural variation in patient genomes using nanopore sequencing. Nat Commun. 8(1):1326 (2017).






