Blog: Ruminating on the rumen microbiome
In this blog, Beatriz Delgado Corrales, Óscar González Recio, and Mónica Gutiérrez Rivas describe their work on classifying rumen microbiomes, comparing long-read and short-read sequencing platforms.
The rumen microbiome has been the subject of study for decades, due to the economic value and environmental impact of dairy cattle in our society (Knapp et al., 2014). Previous studies of the rumen composition have been based either on traditional microbiology techniques, such as microbial cultures, or 16S rRNA analysis, which is widely used for taxonomic profiling (Seshadri et al., 2018). However, microbial cultures are usually time-consuming, difficult, and don’t provide full information about the microbial composition (Creevey et al., 2014), while 16S rRNA analysis, which only accounts for certain regions of the 16S rRNA gene, can introduce bias and does not provide accurate taxonomic classification (Knight et al., 2018).
Benefits of whole-genome sequencing vs. targeted analysis of microbiomes
An alternative to cultures and 16S rRNA sequencing is whole genome sequencing and assembly, which provides a better taxonomic classification based on multiple marker genes, and does not introduce PCR biases. Short-read sequencing is still considered the gold standard for microbiome studies, but it is expensive, non-portable and requires long protocols. Moreover, short-read sequencing still faces multiple challenges, including the difficulty in assembling full genomes or repetitive regions that can hold important genomic information (Quince et al., 2017). Nanopore sequencing with MinION or GridION can be used to provide better assemblies by increasing the read length with no technical limit, and provides rapid, cost-effective and on-site sequencing, allowing animal studies and veterinary diagnostics to be performed in the field.
A comparison of sequencing technologies
In our recent publication (Delgado, Serrano, González, Bach, & González-Recio, 2019), we compared short-read to MinION sequencing to determine the accuracy of the microbial profiling between the two. To do so, we sequenced the rumen content of 12 cows that were selected for extreme feed efficiency phenotype using the MinION and Illumina MiSeq. For nanopore sequencing, we used the Ligation Sequencing Kit (SQK-LSK109 for 1D metabarcoding) for whole genome sequencing of the samples. While we initially tried the Rapid Sequencing Kit, we achieved better performance with SQK-LSK109, with less pore saturation, longer runs, and higher output.
To compare both outputs, we followed two different pipelines: (1) assembly-free, which provides a quick taxonomic profiling of the microbiome, and (2) assembly-based, which allows a better insight of the microbial genomes for functional annotation and genome analysis. For the assembly-free approach, we used DIAMOND for protein blast against the NCBI database, and MEGAN for taxonomic profiling and visualization. For the assembly-based approach we used Megahit to assemble the short reads and Canu for nanopore reads.
Nanopore sequencing is an easier, faster approach for microbiome profiling
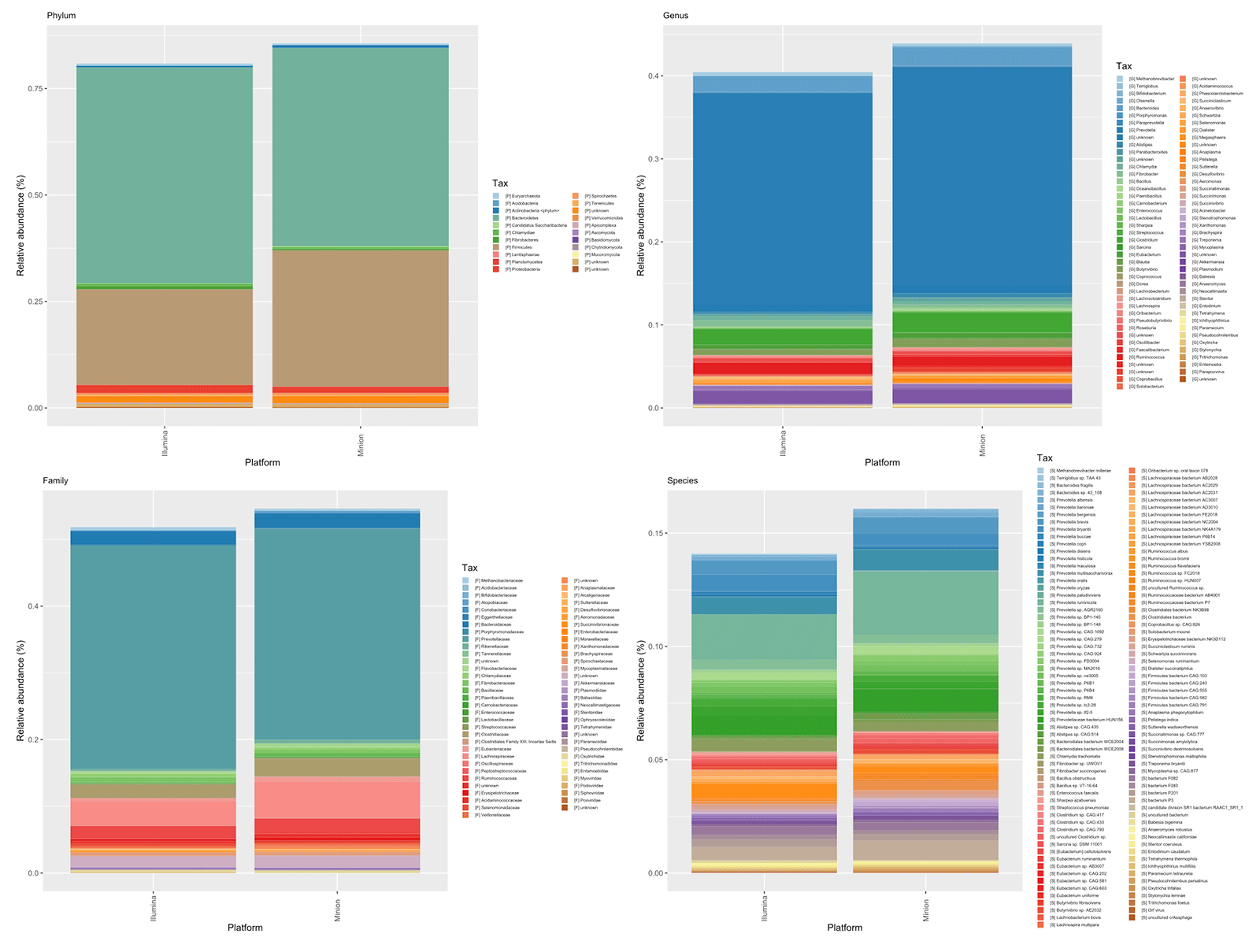
Overall, our analyses suggest that nanopore sequencing provides an interesting alternative for microbiome profiling in rumen samples following this protocol. The microbiome compositions determined using the two technologies are similar (Figure 1), especially at the phylum and family levels, although a larger number of taxonomic groups are detected when using longer reads. However, low-abundancy species like some methanogens (Methanobrevibacter spp.) were not detected by nanopore reads, likely due to its low sequencing depth compared to short-read sequencing methods. To overcome this problem, more efforts should be made to increase the number of sequenced reads to have a better insight of relevant species in the rumen.
The assembly using only nanopore reads provided fewer but longer assembled contigs (Table 1), which is very useful for analysis of whole microbial genomes. However, the sequencing depth plays an important role in assemblies, and deep sequencing protocols need to be developed for nanopore sequencing of the rumen microbiome. This can involve using less aggressive DNA extraction kits to obtain longer DNA fragments or eliminating the host DNA before sequencing.
| Illumina assembly | MinION assembly | |
|---|---|---|
| Length (bp) | 5,296,436 | 2,550,082 |
| GC (%) | 47.7 | 46.89 |
| N's | 0 | 0 |
| N50 | 641 | 11,549 |
| Largest Contig (bp) | 11,173 | 49,178 |
| Contigs | 7,856 | 281 |
Table 1. Assembly statistics for both types of sequencing
We determined that nanopore sequencing using the MinION provides an easier approach to microbiome profiling for almost all kinds of laboratories due to its small size, flexibility, and relatively low cost. Oxford Nanopore provides us with flexibility to analyse microbiome profiles in real time and provides quick results that may be used for in-field identification or phenotyping in dairy cattle. We have already sequenced the whole rumen metagenome of over 800 samples.

References
- Creevey, C. J., Kelly, W. J., Henderson, G., & Leahy, S. C. (2014). Determining the culturability of the rumen bacterial microbiome. Microbial Biotechnology, 7(5): 467–479.
- Delgado, B., Serrano, M., González, C., Bach, A., & González-Recio, O. (2019). Long reads from Nanopore sequencing as a tool for animal microbiome studies. BioRxiv, DOI: https://doi.org/10.1101/2019.12.21.886028.
- Knapp, J. R. et al. (2014). Invited review: Enteric methane in dairy cattle production: Quantifying the opportunities and impact of reducing emissions. Journal of Dairy Science, 97(6): 3231-3261.
- Knight, R. et al. (2018). Best practices for analysing microbiomes. Nature Reviews Microbiology, 16: 410-422.
- Quince, C. et al. (2017). Shotgun metagenomics, from sampling to analysis. Nature Biotechnology, 35: 833–844.
- Seshadri, R. et al. (2018). Cultivation and sequencing of rumen microbiome members from the Hungate1000 Collection. Nature Biotechnology, 36: 359–367.
 Beatriz Delgado CorralesI am a biotechnologist, at the University of Queensland, specialized in environmental microbiology through the Autonomous University of Madrid (UAM). I conducted my Bachelor’s and Master’s thesis at the National Institute for Agricultura and Food Research (INIA), on the rumen microbiome, its impact on methane emissions, and relevance in animal breeding and genetics; first, by studying the production of methane by methanogenic archaea, and later doing metagenomic analysis of the rumen microbiome using short-read and nanopore sequencing with the MinION. I am now focused on applying different bioinformatic pipelines to analyze metagenomes and recover assembled microbial and viral genomes and predict their metabolic capacities in alkane-rich environments.
Beatriz Delgado CorralesI am a biotechnologist, at the University of Queensland, specialized in environmental microbiology through the Autonomous University of Madrid (UAM). I conducted my Bachelor’s and Master’s thesis at the National Institute for Agricultura and Food Research (INIA), on the rumen microbiome, its impact on methane emissions, and relevance in animal breeding and genetics; first, by studying the production of methane by methanogenic archaea, and later doing metagenomic analysis of the rumen microbiome using short-read and nanopore sequencing with the MinION. I am now focused on applying different bioinformatic pipelines to analyze metagenomes and recover assembled microbial and viral genomes and predict their metabolic capacities in alkane-rich environments.  Óscar González RecioI am a Senior Research Scientist at the National Institute for Agricultural and Food Research in Spain. I am also associate professor at the Polytechnic University of Madrid. I have been working in the statistical genomics field to investigate aspects related to fertility, genomic selection and epigenetics in livestock. Since 2013, my research mainly focuses on genomic applications to breeding more efficient and sustainable livestock with lower methane emissions. The role of the microbiome in the sustainability of animal husbandry is of key importance, and the use of metagenomic information in animal breeding has been getting a lot of attention in recent years. We aim to characterize the rumen microbiota and determine the microbes that confer a more efficient digestion of feed in ruminants. The MinION device will allow us to provide a quick determination of feed efficiency for farms and even in situ.
Óscar González RecioI am a Senior Research Scientist at the National Institute for Agricultural and Food Research in Spain. I am also associate professor at the Polytechnic University of Madrid. I have been working in the statistical genomics field to investigate aspects related to fertility, genomic selection and epigenetics in livestock. Since 2013, my research mainly focuses on genomic applications to breeding more efficient and sustainable livestock with lower methane emissions. The role of the microbiome in the sustainability of animal husbandry is of key importance, and the use of metagenomic information in animal breeding has been getting a lot of attention in recent years. We aim to characterize the rumen microbiota and determine the microbes that confer a more efficient digestion of feed in ruminants. The MinION device will allow us to provide a quick determination of feed efficiency for farms and even in situ.  Mónica Gutiérrez RivasI have been studying RNA viruses since 1997: during my PhD at Esteban Domingo´s lab at CBMSO (Centro de Biología Molecular Severo Ochoa) I focused my research on the quasispecies distribution of RNA viruses, the dynamics of error-copy production by viral polymerases and their error threshold and lethal mutagenesis relationships. Since then, I have been focusing on the implications of viral quasispecies in tropism changes, antiviral and immune resistance, conducted both at human and animal research centers, CNM-ISCIII and CISA-INIA, respectively. I am currently part of the METALGEN project, under Oscar Gonzalez Recio´s supervision, at INIA, to analyze cow genetics, methane emissions, rumen microbiomes, and their relationships. We are interested in applying nanopore sequencing to the identification of the rumen microbiome, using the MinION device.
Mónica Gutiérrez RivasI have been studying RNA viruses since 1997: during my PhD at Esteban Domingo´s lab at CBMSO (Centro de Biología Molecular Severo Ochoa) I focused my research on the quasispecies distribution of RNA viruses, the dynamics of error-copy production by viral polymerases and their error threshold and lethal mutagenesis relationships. Since then, I have been focusing on the implications of viral quasispecies in tropism changes, antiviral and immune resistance, conducted both at human and animal research centers, CNM-ISCIII and CISA-INIA, respectively. I am currently part of the METALGEN project, under Oscar Gonzalez Recio´s supervision, at INIA, to analyze cow genetics, methane emissions, rumen microbiomes, and their relationships. We are interested in applying nanopore sequencing to the identification of the rumen microbiome, using the MinION device. 





