Blog: Resolving structural variants causing antithrombin deficiency
In this blog, Alba Sanchis-Juan, Javier Corral, and Belén de la Morena-Barrio describe their research into the genetic basis of thrombophilia, and how nanopore long sequencing reads were needed to resolve the structural variants found to be involved.
Structural variants (SVs) are genomic rearrangements that contribute to genomic diversity, function, and evolution, and can cause somatic and germline diseases (Sudmant et al, 2015). However, the identification and characterization of SVs in clinical genetics have remained historically challenging as routine genetic diagnostic techniques have limited ability to evaluate repetitive regions and SVs. These limitations may now be addressed by long-read sequencing technologies such as nanopore sequencing (Sanchis-Juan et al, 2018).
In our recent publication (De la Morena-Barrio et al, 2020), we have used nanopore sequencing to resolve SVs involved in antithrombin deficiency type I (ATD). ATD is the most severe thrombophilia that significantly increases the risk of venous thrombosis (Corral et al, 2018) and it is caused by haploinsufficiency of SERPINC1 gene. Despite being the first thrombophilia described 55 years ago and having more than 440 different causal variants identified, up to 30% of cases with ATD remain unresolved. Additionally, the high number of repetitive elements in and around SERPINC1 challenges the identification of SVs by routine diagnostic methods (Corral et al, 2018).
Our nanopore sequencing-based approach
Nanopore sequencing presents a powerful approach to overcome these limitations, since the long reads can span the repetitive regions, allowing identification and characterization of SVs at nucleotide level resolution. In our study, we performed nanopore sequencing using the PromethION platform on 19 unrelated individuals with ATD, where routine molecular tests were either negative, ambiguous, or not fully characterized, in order to identify, resolve and investigate the most likely molecular mechanism of formation of causal SVs involved in this severe thrombophilia (Figure 1A).
We performed 21 runs to reach desired coverage, using the 1D Ligation Sequencing library prep kit, and sequencing with R9 flow cells. The average median genome coverage obtained was 16x (sd ± 7.7) and the average read length was 4,499 bp (sd ± 4,268), although very long reads were also obtained until a maximum of 2.5 Mbp (Figure 1B). Our multi-modal analysis workflow was applied to all the samples for the sensitive detection of SVs, and is publicly available here.
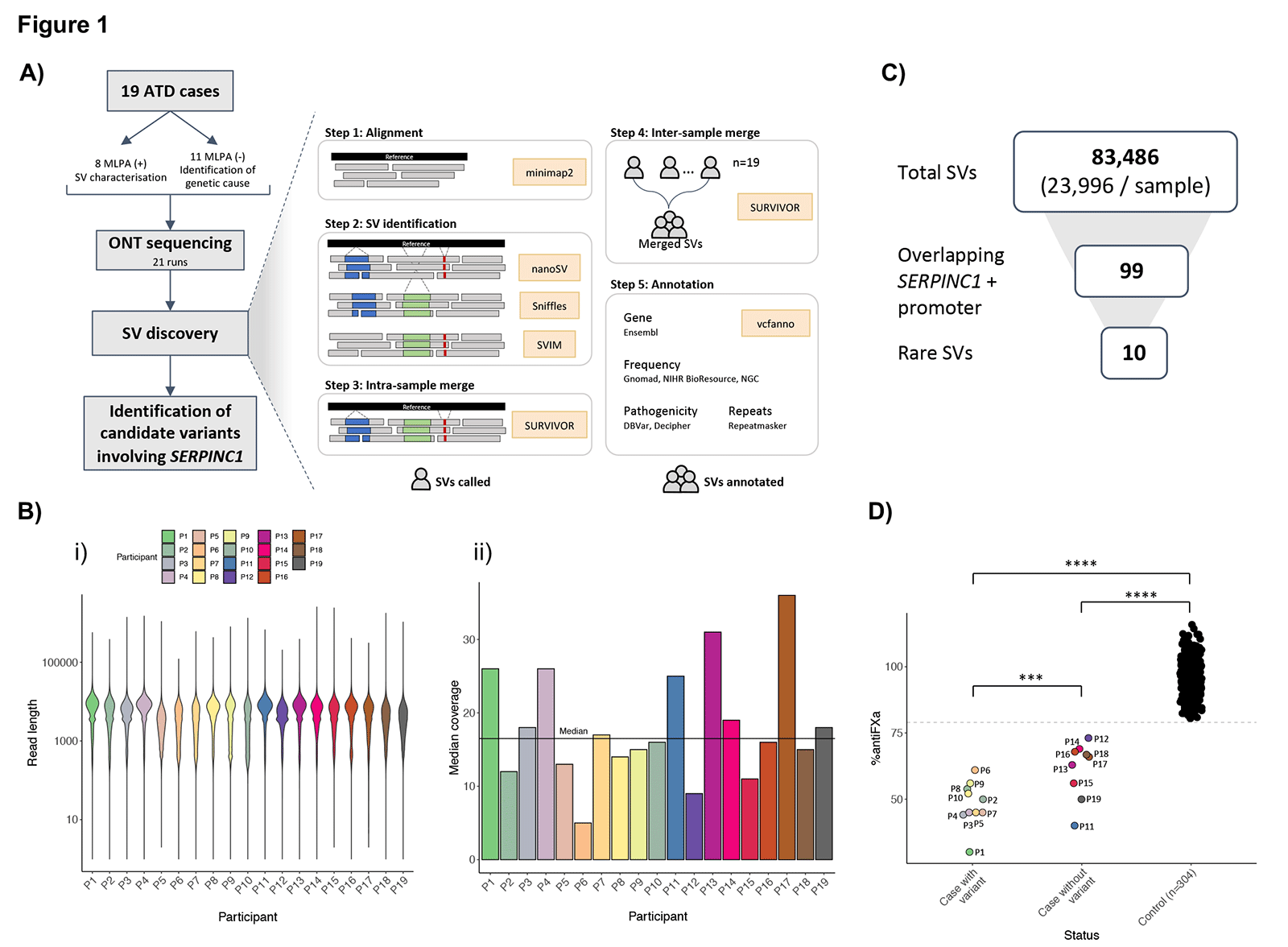
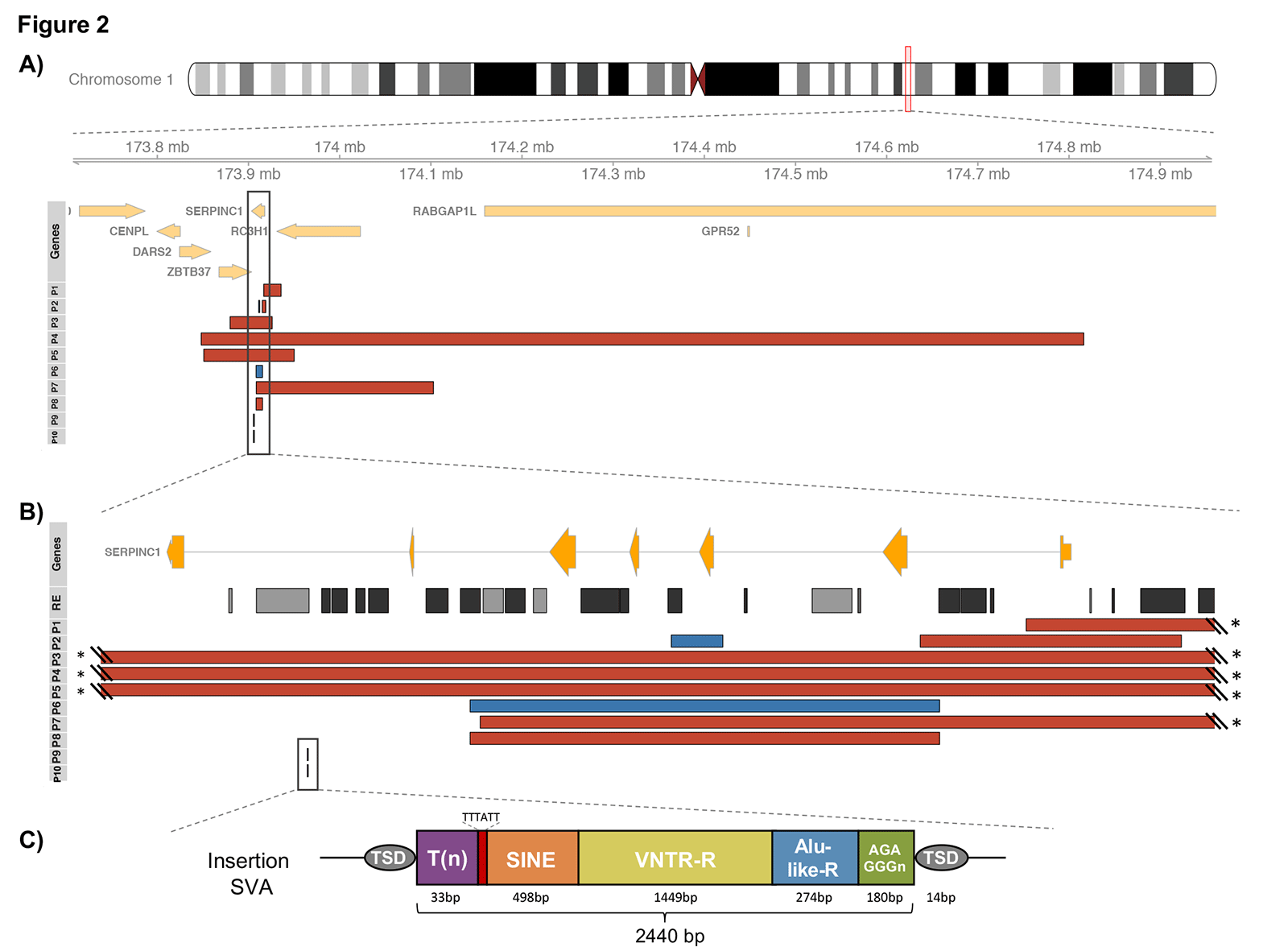
Disease-associated SVs affecting SERPINC1 were identified for 10 cases, and varied in size (from 7 Kbp to 1 Mbp) and type (six deletions, one duplication, one complex SV, and two large insertions) (Figures 1C, 1D and 2). Our study resolved ambiguous SVs, and, more importantly, we identified for the first time a complex germline rearrangement involved in ATD, previously misclassified by routine diagnostic methods as a deletion.
Remarkably, we also revealed the molecular basis of two unrelated cases with previously unknown genetic defect(s); they harboured the insertion of a novel SINE-VNTR-Alu (SVA) retroelement in an intron of SERPINC1 (Figure 2C), which was characterised by de novo assembly and confirmed by specific PCR amplification in other affected family members. This is the first report showing this mechanism as causative of ATD, and enlarges the panel of disorders where SVA retroelements are involved.
Nanopore sequencing facilitated breakpoint analysis, revealing the presence of repetitive elements in all the SVs, Alu elements being the most frequent and involved in some instances with a non-random formation. Additionally, microhomologies, small insertions, deletions and/or duplications were also observed for most of the SVs, suggesting a replication-based mechanism (such as BIR/MMBIR/FoSTeS) for the generation of these SVs.
Conclusions and outlook
Overall, we resolved SVs in 10 individuals. However, there are still cases with ATD that remain unresolved. In future studies we plan to evaluate epigenetic mechanisms, regulatory defects, and variations in other genes that might cause ATD. All these analyses will also be explored by nanopore sequencing, alone or in combination with other technologies.
Our results highlight the important advantages that nanopore sequencing presents over alternative sequencing methods for resolving, identifying, and unveiling the molecular mechanisms of formation of disease-causing SVs. We recommend its use as a complementary method to investigate causality of ATD and other congenital disorders, especially when SVs are suspected to be involved.
References
- Sudmant, P., Rausch, T., Gardner, E. et al. An integrated map of structural variation in 2,504 human genomes. Nature. 526, 75–81 (2015).
- Sanchis-Juan, A., Stephens, J., French, C.E. et al. Complex structural variants in Mendelian disorders: identification and breakpoint resolution using short- and long-read genome sequencing. Genome Med. 10, 95 (2018).
- Corral J., de la Morena-Barrio M.E., Vicente V. The genetics of antithrombin. Thromb Res. 169:23-9 (2018).
- de la Morena-Barrio B., Stephens J., de la Morena-Barrio M.E., et al. Long-read sequencing resolves structural variants in SERPINC1 causing antithrombin deficiency and identifies a complex rearrangement and a retrotransposon insertion not characterized by routine diagnostic methods. bioRxiv. 2020.08.28.271932 (2020).
 Alba Sanchis-JuanAlba studied Biochemistry and Biomedicine, and did her PhD in Biotechnology at the University of Valencia, Spain. She worked for four years at the Department of Haematology, University of Cambridge, as part of the NIHR Bioresource Project, where she focused on the discovery of unknown etiological genes and variants in coding and non-coding regions of the genomes of patients with rare diseases. Fascinated by the complexity of the human genome, she worked on identifying and characterizing causal structural variants using multiple sequencing technologies, including short- and long-read whole-genome sequencing. Currently, she is a Postdoctoral Fellow at the Talkowski laboratory in the MGH, Broad Institute and Harvard Medical School, where she works on large-scale population and clinical genomics projects to systematically explore a variety of genomic variation and its implication for human disease.
Alba Sanchis-JuanAlba studied Biochemistry and Biomedicine, and did her PhD in Biotechnology at the University of Valencia, Spain. She worked for four years at the Department of Haematology, University of Cambridge, as part of the NIHR Bioresource Project, where she focused on the discovery of unknown etiological genes and variants in coding and non-coding regions of the genomes of patients with rare diseases. Fascinated by the complexity of the human genome, she worked on identifying and characterizing causal structural variants using multiple sequencing technologies, including short- and long-read whole-genome sequencing. Currently, she is a Postdoctoral Fellow at the Talkowski laboratory in the MGH, Broad Institute and Harvard Medical School, where she works on large-scale population and clinical genomics projects to systematically explore a variety of genomic variation and its implication for human disease.  Javier CorralJavier Corral is an Associate Professor of Experimental Hematology at the University of Murcia, Spain. He obtained his PhD in Biochemistry and Molecular Biology at the University of Salamanca (1992), and was awarded a Postdoctoral Fellowship at the Medical Research Council Laboratory of Molecular Biology (MRC-LMB)/ PNAC Division, and was Fellow of Trinity College and scientific visitor at the Welcome Trust Centre, CIMR, University of Cambridge (UK). Professor Corral's research interests cover the area of thrombosis, specifically antithrombin, a key endogenous anticoagulant. Dr. Corral has authored 193 articles in peer-reviewed journals such as Blood, Circulation, PNAS, Cell, EMBO, Nature Genetics, Haematologica, and the Journal of Thrombosis and Haemostasis. Dr. Corral has been vice-president of the Spanish Society of Thrombosis and Haemostasis, and is member of the editorial board of several journals.
Javier CorralJavier Corral is an Associate Professor of Experimental Hematology at the University of Murcia, Spain. He obtained his PhD in Biochemistry and Molecular Biology at the University of Salamanca (1992), and was awarded a Postdoctoral Fellowship at the Medical Research Council Laboratory of Molecular Biology (MRC-LMB)/ PNAC Division, and was Fellow of Trinity College and scientific visitor at the Welcome Trust Centre, CIMR, University of Cambridge (UK). Professor Corral's research interests cover the area of thrombosis, specifically antithrombin, a key endogenous anticoagulant. Dr. Corral has authored 193 articles in peer-reviewed journals such as Blood, Circulation, PNAS, Cell, EMBO, Nature Genetics, Haematologica, and the Journal of Thrombosis and Haemostasis. Dr. Corral has been vice-president of the Spanish Society of Thrombosis and Haemostasis, and is member of the editorial board of several journals.  Belén de la Morena-BarrioBelén de la Morena-Barrio graduated in Pharmacy in 2015. Currently, she is doing her PhD in the group of Prof. Javier Corral at the University of Murcia, Spain. Her research is focused on the identification of new molecular mechanisms involved in antithrombin deficiency by evaluating cases with unknown molecular causes, and characterizing structural variants involved in this disorder. She is an expert in 3rd Generation Sequencing, thanks to her training in the group of Prof. Willem Ouwehand at the University of Cambridge. At three years into her PhD, she has published 8 articles, being the first author of 3 of them.
Belén de la Morena-BarrioBelén de la Morena-Barrio graduated in Pharmacy in 2015. Currently, she is doing her PhD in the group of Prof. Javier Corral at the University of Murcia, Spain. Her research is focused on the identification of new molecular mechanisms involved in antithrombin deficiency by evaluating cases with unknown molecular causes, and characterizing structural variants involved in this disorder. She is an expert in 3rd Generation Sequencing, thanks to her training in the group of Prof. Willem Ouwehand at the University of Cambridge. At three years into her PhD, she has published 8 articles, being the first author of 3 of them. 





