Accurate poly(A) tail length estimation for Direct RNA Sequencing (SQK-RNA002) and cDNA (SQK-PCS111, SQK-PCB111.24) sequencing kits

Requirements
Accurate poly(A) tail length estimation for Direct RNA Sequencing (SQK-RNA002) and cDNA (SQK-PCS111, SQK-PCB111.24) sequencing kits
FOR RESEARCH USE ONLY.
Contents
Estimating poly(A) tail lengths for known standards
Change log
Many RNA transcripts, including some viral RNA genomes and long, non-coding RNA (lncRNA), have non-template homopolymers of adenosine added to the 3’ end of the nascent transcript during transcriptional processing within the nucleus. Most notably, eukaryotic messenger RNA (mRNA) transcripts have homopolymer adenosine, “poly(A)”, tails that can be over 200 nucleotides in length, depending on the organism or other influencing factors. These poly(A) tails are important in the life cycle of an RNA transcript for many processes, such as transportation out of the nucleus, translation regulation, long-term storage during quiescence, and stability from degradation. Eukaryotic mRNA transcript poly(A) tail lengths can alter in length throughout its life cycle – both decreasing and increasing – providing insight into the different cellular processes acting upon it. Therefore, it is useful to have the means to measure poly(A) tail lengths accurately and robustly. Previously, poly(A) tail lengths were frequently measured through non-sequencing methods, such as blotting and/or radioactive probing, or the tails were sequenced but information pertaining to the associated body of the transcript was not included. The Oxford Nanopore Technologies' Direct RNA Sequencing (SQK-RNA002) and cDNA (SQK-PCS111 and SQK-PCB111.24) kits are the only sequencing kits, at present, by which it is possible to capture complete transcript information though sequencing full-length RNA transcripts and accurately estimating the length of poly(A) tails associated with them. There are several poly(A) tail length estimation software pipelines available for the Direct RNA Sequencing Kit (SQK-RNA002) data. Two of which we recommend are nanopolish and tailfindr. For cDNA (SQK-PCS111 and SQK-PCB111.24) data, it is possible to estimate poly(A) tail lengths using tailfindr.
Note:
- For both nanopolish and tailfindr, it is necessary to collect .fast5 data files for downstream analyses in addition to .fastq files. Ensure the sequencing parameters of your experiment in MinKNOW are set to collect both file types before you start sequencing and that you have enough data storage available for collecting numerous .fast5 files.
- To use Tailfindr, the .fast5 data needs to be re-basecalled using standalone Guppy (version 6.3.8 or older), including the
--fast5_outoption in the basecalling command. This will generate a new set of .fast5 files with the basecalling information included and to generate move and trace tables.
Estimating poly(A) tail lengths for known standards
Experiment I.
In vitro transcribed (IVT) RNA transcripts for seven unique Escherichia coli- and two Saccharomyces cerevisiae-derived genes were generated, each with a defined number of homopolymeric adenosines incorporated downstream of the gene. The poly(A) tails in each of the IVT transcripts was engineered as a specific length of 10-100 adenosine nucleotides with respect to the transcript gene body (Table 1).
 Table 1. In-vitro transcribed transcript standards with defined poly(A) tail lengths.
Table 1. In-vitro transcribed transcript standards with defined poly(A) tail lengths.
cDNA-PCR libraries (SQK-PCS111) were prepped for each of the seven E. coli-derived IVT transcripts using 2 ng of input material and pooled before loading 25 ng of mixed library on duplicate flow cells. Additionally, direct RNA libraries (SQK-RNA002) were prepared with 70 ng starting input amount and pooled before loading on duplicate flow cells. The samples were run according to their respective kits, with .fast5 files collected, and the data for each of the duplicate runs were combined respective to each kit.
Poly(A) tails for both direct RNA and cDNA libraries were estimated using tailfindr for both kits and nanopolish for direct RNA (Figure 1). For direct RNA, the median observed tail length corresponds to the expected tail lengths for each of the standards when using both tail estimation methods (Figure 1A). The PCR amplified cDNA libraries can be sequenced bidirectionally with respect to transcript orientation due to the sample preparation design, therefore, the poly(dA) and poly(dT) lengths corresponding to the transcript poly(A) tail were measured using tailfindr (Figure 1B). Again, the median observed poly(A) tail lengths for both orientations correspond to the expected length with no significant bias. The wider distribution of tail length estimations observed for the individual IVT transcripts containing 70 and 100 adenosines is likely due to the sample containing a range of polyadenylation lengths, possibly due to RNA degradation or transcription artefacts, and not the tail length estimation methods (Figure 1).
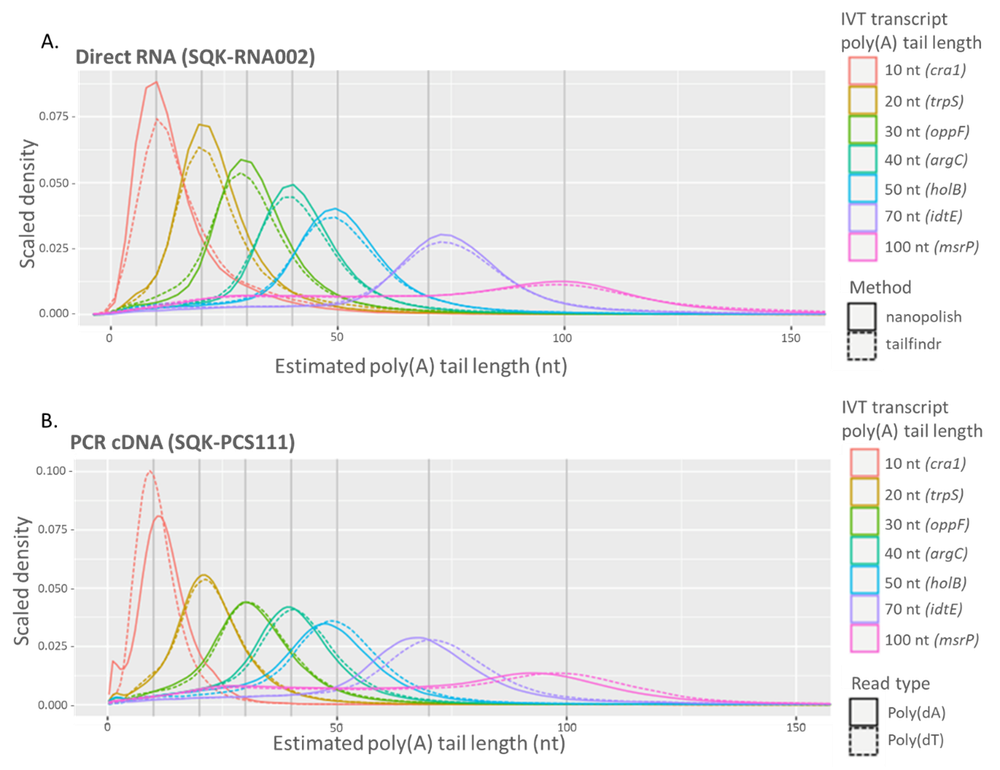
Figure 1. Poly(A) tail estimates for transcripts with known poly(A) tail lengths of 10-100 adenosine nucleotides. A) Tail lengths for direct RNA (SQK-RNA002) libraries were estimated using nanopolish (solid line) and tailfindr (dashed line). B) Tail lengths were estimated from cDNA (SQK-PCS111) libraries from both the sense (solid line) and antisense (dashed line) using tailfindr.
Experiment II.
We spiked two yeast-derived IVT transcripts with known poly(A) tail lengths of 30 or 100 homopolymer adenosines into a background of ribodepleted, enzymatically polyadenylated E. coli total RNA and prepped both direct RNA and cDNA libraries (Table 1). Additionally, these standards were spiked into the negative control preps of E. coli total RNA, non-polyadenylated direct RNA and cDNA libraries. More information regarding libraries for this experiment can be found in our 'Polyadenylation of non-poly(A) transcript using E. coli poly(A) polymerase' document. The yeast-derived IVT spike-ins comprised of 1% of total reads and their tail lengths were measured using tailfindr to compare the direct RNA and cDNA libraries.
The median tail length observed for both IVT transcripts in the direct RNA and the cDNA libraries correlate to the expected lengths for each, respectively (Figure 2 and 3). The distribution of estimated tail lengths is narrower for the yeast-derived Eno2 transcript (Figure 2B and 3B) than the E. coli-derived msrP IVT transcript, each with a putative poly(A) tail of 100 adenosines (Figure 1). This further suggests that a wider tail length estimate distribution is indicative of the sample and not the tail length estimation methodology. It is worth noting that we do observe a slight overestimation for the Eno2 transcript poly(A) tail in the cDNA libraries (Figure 3B) when compared to the direct RNA libraries (Figure 2B). For best practice when determining exact lengths, we recommend prepping both direct RNA and cDNA libraries and comparing them using both nanopolish and tailfindr where possible.
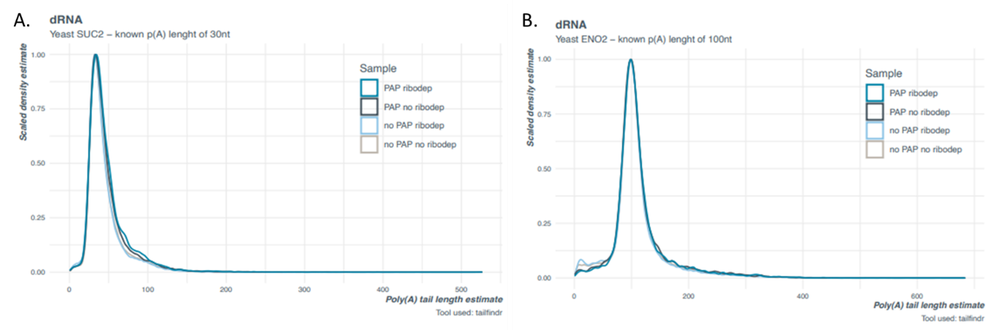
Figure 2. Tail length estimates for yeast-derived IVT transcripts spiked into biological samples of enzymatically polyadenylated E. coli direct RNA (SQK-RNA002) libraries using tailfindr. A) Suc2 with 30 nt poly(A) tail and B) Eno2 with 100 nt poly(A) tail.
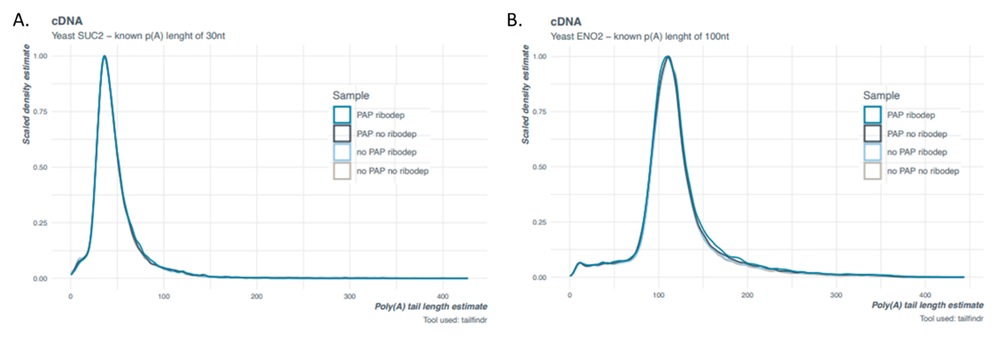
Figure 3. Tail length estimates for yeast-derived IVT transcripts spiked-into biological samples of enzymatically polyadenylated E. coli cDNA (SQK-PCS111) libraries using tailfindr. A) Suc2 IVT transcript with 30 nt poly(A) tail and B) Eno2 IVT transcript with 100 nt poly(A) tail.
Change log
| Version | Change note |
|---|---|
| V2, April 2023 | Updated recommendations for using tailfindr |
| V1, October 2022 | Initial protocol publication |






