Leaving the lab bench: on-farm genotyping in remote Australian cattle farms
What is distinct about Australian cattle farms?
Australian cattle farms are enormous. These large properties span thousands of square kilometres and some are larger even than small countries like Israel and Wales. Often located in some of the most remote regions in the world, major cities can be up to a 12 hour drive away. Due to the extensive size of these farms they are largely unfenced, and cattle are only handled once a year for animal husbandry and management purposes. This process of finding and yarding the cattle is known as ‘mustering’. To find the cattle on these large farms, helicopters, small aeroplanes, motorbikes, and horses are used, making mustering an expensive activity.

Genetics in the livestock industry and challenges faced without onsite sequencing
A useful management tool employed by farmers around the world is genotyping. Genotyping of cattle allows for the selection of animals with increased genetic merit1. This allows producers to breed for important production traits, such as feed conversion and fertility, which ultimately increase productivity and decrease greenhouse gas emissions from cattle. This type of genomic selection has also been used to improve animal welfare by selecting for genetic mutations that confer desirable traits such as calmer temperament. A typical genomic selection workflow involves mustering animals to take DNA samples, sending the samples to a genetics lab, waiting for the results, and re-mustering animals once results are ready. The results are then used to sort animals according to their genetic potential. Presently, turnaround times for genotyping in Australia range between 6–8 weeks. Unfortunately, due to the prohibitive cost of mustering on Australia’s large cattle farms, results from genotyping currently can’t be implemented until the following year. This makes improving the genetics of a herd on Australian cattle farms extremely slow. Therefore, a rapid genotyping alternative is necessary to accelerate this process in Australia’s cattle industry and increase productivity.
Introducing on-farm genotyping
Given the need for a rapid genotyping alternative, we (an agrigenomics group at the University of Queensland) decided to investigate the feasibility of genotyping animals on-farm using Oxford Nanopore Technologies’ portable sequencing device, the MinION. Key to the feasibility of this method are accuracy, turnaround time (less than 48 hours), and cost (less than USD $40/animal). This means the ability to multiplex a high number of samples on each flow cell is essential.

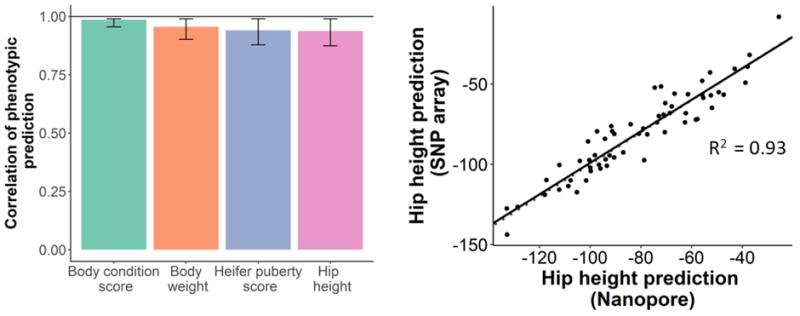
Our work began by benchmarking the accuracy of existing genotyping technologies, such as low-density SNP arrays, against different ultra-low coverage genotyping methods using nanopore sequencing2. Four different low-coverage genotyping methods were evaluated across five sequencing coverages from 0.05x to 2x. We found that by using QUILT3 for genotyping at sequencing coverages as low as 0.1x, we achieved an average genotyping accuracy of 89% and similar genomic predictions accuracies to those possible with commercial low-density SNP arrays. With a genome size of 2.7 Gb, this means only 0.27 Gb of data per animal is needed for accurate prediction of important phenotypes.
Why not ask more?
A major benefit of employing sequencing over SNP array genotyping, is the ability to capture a greater range of genetic information from a single sample. For example, when genotyping tail hair samples from cattle, SNP arrays are only able to test pre-defined SNP markers, whereas sequencing detects all DNA present in the sample, including viral and bacterial DNA. Recognising the potential to use this flexibility to offer enhanced information to farmers, we decided to investigate whether we could detect bacteria and viruses from the same sample while genotyping animals. By rolling disease testing and genotyping into a single test, we hoped to increase the value proposition of on-farm genotyping.
To test this hypothesis, we genotyped animals from nasal swabs rather than tail hair4. We hoped that the nasal swabs would provide the right balance between host and non-host DNA to allow for accurate prediction of phenotypic traits as well as accurate profiling of respiratory pathogens. Nasal swabs from 48 animals presenting with symptoms of bovine respiratory disease were multiplexed on a PromethION Flow Cell. After 48 hours of sequencing, traits were predicted with an average correlation of 0.92. KRAKEN5 was used for taxonomic classification of contaminant reads and the results were compared to qPCR results for common bovine respiratory disease pathogens from the same sample. Viral and bacterial pathogens related to bovine respiratory disease were detected in all samples and were correlated with the qPCR results. This research suggests we are able to provide rapid phenotypic trait predictions and disease diagnosis for farmers from the same sample. Importantly, at this level of multiplexing, the cost of consumables was USD $22 per sample; however, this does not include the cost of labour.
A limitation of our ability to implement this research on-farm is the throughput capacity of the MinION sequencer. The release of the PromethION 2 has significantly increased the potential throughput of this method. This advancement, in combination with the increasing throughput of flow cells and improvements in basecalling accuracy, will continue to decrease the price of nanopore skim genotyping-by-sequencing, making it a feasible solution for on-farm genotyping and pathogen detection.

1. Hayes B., Goddard M. Genome-Wide Association and Genomic Selection in Animal Breeding. Genome. 53:876-83 (2010).
2. Lamb H.J. et al. Imputation Strategies for Genomic Prediction Using Nanopore Sequencing. BMC Biol. 21:286 (2023).
3. Davies R.W. et al. Rapid Genotype Imputation from Sequence with Reference Panels. Nature Genetics. 53:1104-1111 (2021).
4. Lamb H.J. et al. Skim-Nanopore Sequencing for Routine Genomic Evaluation and Bacterial Pathogen Detection in Cattle. Animal Production Science. 63:1074-1085 (2023).
5. Lu J. et al. Metagenome Analysis Using the Kraken Software Suite. Nature Protocols. 17:2815-2839(2022).






