Nanopore Community Meeting - Day 2 writeup
Gordon Sanghera opened Day 2 to Blondie, of course, and by announcing that two service providers have now completed certification and are offering nanopore sequencing as a service using GridION. read more here:

Plenary session - Patricia Simner
Coming soon! In the meantime, you can watch the post-plenary interview:

Lightning talks
Tara Alpert
Tara Alpert from Yale School of Medicine showcased her work utilising nanopore sequencing to investigate gene-specific variation of co-transcriptional splicing efficiency. In many organisms, splicing occurs co-transcriptionally (i.e. before transcription terminates) and the interplay between RNA polymerase II and the spliceosome has an important role in regulating gene expression. Mutations in in these mechanisms are found to be highly prevalent in cancer and other genetic disorders.
Using budding yeast as a model, the team set out to understand the gene factors that influence splicing. To find out how fast splicing occurs, they developed a targeted sequencing approach called Single Molecule Intron Tracking (SMIT). This helps to determine the position of RNA polymerase II along the gene when splicing occurs for individual transcripts.
Initial work utilising another long read technology delivered too few reads, so they are now recapitulating this experiment using nanopore sequencing. Tara believed that this will eliminate library prep steps that introduce bias, reduce cost and speed up data collection. The long read lengths will also facilitate the team to study other, larger organisims.
Lee Kerkhof
The microbiome is known to play an important role in host health, in this second lighting talk, Lee Kerkhof from Rutgers University discussed his research into how host genetics, behaviour and exposure to toxins alters the microbiome. Lee presented the results of his initial work assessing the impact of ozone treatment on mouse lung microbiome composition. Using the MinION, Lee was able to rapidly detect significant growth of opportunistic pathogens in ozone exposed mice.
The team at Rutgers also want to understand the interaction between the host and microbes. To do this, they utilised 13C amino acids and stable isotope probing (SIP) in order to distinguish active bacteria utilising the carbon source. The data matches well with the opportunistic pathogens that grow after ozone exposure.
Andrew Routh & Elizabeth Jaworski
Andrew Routh and Elizabeth Jaworski from the University of Texas Medical Branch at Galveston showcased his team’s research characterising the evolution of defective interfering (DI) RNA viruses using nanopore sequencing. DI RNAs are copies of RNA viruses that occur naturally during viral infections but have been truncated and rearranged by recombination. They play important roles in viral pathology as they can attenuate viral replication, alter the outcome of viral infection, induce viral persistence, and have been proposed as anti-viral therapeutics.
Andrew presented data on their recent study utilising a combination of long- and short-read sequencing to characterise the evolution of DI RNAs during passaging of Flock House virus in cell-culture. This combined approach overcomes the limitations of short-read sequencing, which is unable to provide gene linkage information. The data allowed the team to elucidate the rapid evolution of DI, with ‘mature’ DI RNAs characterised by multiple recombination events. Andrew suggested that nanopore sequencing provides a unique platform to study the sequencing topology, evolution and abundance of DI RNAs. The team now plan to characterise the formation and evolution of DI RNAs in clinical isolates.

Michael Bartsch
Michael Bartsch of Sandia National Laboratories presented RUBRIC (Read Until with Basecall- and Reference Informed Criteria), which builds upon the Read Until selective-sequencing approach developed by Matt Loose’s team at the University of Nottingham. Selective-sequencing, which is unique to nanopore sequencing, allows the user to analyse only DNA strands that contain pre-determined signatures of interest. This is achieved though real-time analysis of the data generated by each individual pore of the flow cell and reversing the voltage of any pore containing unwanted or non-informative sequence. RUBRIC combines real-time basecalling and rapid, on-the-fly alignment to reference sequences as the basis for molecule-by-molecule DNA selection. Furthermore, Michael explained that the system can also be implemented using off-the-shelf PCs as opposed to cluster computing platforms. Michael presented data showing significant depletion of non-target reads, when using RUBRIC to select a specific lambda phage DNA fragment.
Michael concluded by outlining how the RUBRIC selective-sequencing system could have beneficial impact on a number of applications, including point-of-care pathogen diagnostics and metagenomic analysis.

Breakout session: Data analysis - Epigenetics
Alexa McIntyre
N6-methyladenosine (m6A) is a common DNA modification in prokaryotes, which is believed to play a role in bacterial immune systems and host immune evasion. Alexa McIntyre from Weill Cornell presented her recent data comparing the effectiveness of long read direct sequencing technologies for detecting this epigenetic modification across a number of bacterial species.
In order to undertake this research, the team at Weill Cornell developed a new tool, mCaller, which allows detection of m6A from nanopore sequencing data.
Nanopore sequencing was used to confirm known methyltransferase target motifs and validate new motifs detected by an alternative long-read platform, across ten reference species (including Bacillus subtilis, Listeria monocytogenes, Pseudomonas aeruginosa). According to Alexa, the fact that most of the non-motif sites detected by the alternative platform are not detected by nanopore sequencing indicates that these are likely to be false positives in the alternative platform data. Furthermore, the team found that the canonical target motifs that are missed by the alternative platform are identified as methylated using nanopore. They are now extending their studies to include analysis of partially methylated sites. Alexa pointed out that their model is available for other researchers to use and the data will also be released soon.
Marcus Stoiber
Marcus Stoiber, Applications Data Analyst at Oxford Nanopore Technologies, announced the launch of Tombo (tools for non-standard base detection), a new analysis platform that allows detection of epigenetic modifications from raw nanopore sequencing data. Tombo is based on the nanoraw platform; however, a significant difference is that is incorporates k-mer model information, which removes the need for a PCR data set in order to explore modified bases. Marcus explained how Tombo enables researchers to investigate modified bases via three computational methods, providing flexibility with regard to application type and experimental design. The platform allows specific detection of modified bases from a simple to produce samples, enabling straightforward expansion of the catalogue of detectible modifications. In addition, in comparison to nanoraw, the Tombo platform is computationally optimised, delivering faster results and enhanced user experience. Marcus stated that future work will include expansion of the modified base catalogue and further refining the accuracy of detection.
Tombo is freely available to download and is available on GitHub: github.com/nanoporetech/

Scott Gigante
Scott Gigante from Yale University presented his work investigating the epigenetic modifications responsible for differential expression of maternal and paternal alleles in the developing embryo.
Unlike traditional sequencing technology, which requires two separate sequencing reactions and harsh bisulfite treatment to discriminate the DNA modification 5-methylcytosine (5mC), nanopore sequencing allows direct detection of this epigenetic mark alongside sequencing data. Interestingly, Scott explained how bisulfite treatment, which converts unmethylated cytosines to thymine, reduces the complexity of the sequence, thereby reducing the number of discriminating SNPs that can be used to assign alleles to haplotypes (i.e., maternal or paternal origin). Through combining direct sequencing with long read lengths, nanopore sequencing offers the potential for improved haplotype assignment.
Scott compared nanopore sequencing data derived from mouse embryonic placenta tissue with that obtained using matched reduced-representation bisulfite sequencing (RRBS). The nanopore data was analysed using both a novel basecall-based method to haplotype reads and the signal-based nanopolish tool created by Jared Simpson. The results from both methods were pooled together, showing that Albacore’s neural network algorithm gave improved certainty on certain reads compared to the Hidden Markov Model used in nanopolish.
The concordance of the data between the two sequencing platforms confirmed the accuracy of the nanopore-based methylation calls and highlighted the improvement in haplotyping conferred by the longer reads. Scott suggested that, in future, it may be possible to haplotype 90% of their nanopore sequencing reads. Furthermore, taking advantage of longitudinal measurement of methylation along the nanopore reads, Scott reported that the nanopore data allowed identification of novel differentially methylated regions, which due to their proximity to previously unidentified monoallelically expressed genes, suggest possible new imprinting control regions. The team now plan to further investigate these regions and to explore differential methylation in repeat regions that have proven inaccessible to short-read sequencing technologies.
Yunfan Fan
Yunfan Fan from Johns Hopkins University spoke about her work utilising nanopore sequencing to investigate bacterial DNA modifications. Bacteria produce restriction enzymes to destroy viral DNA.In order to prevent digestion of their own DNA, they use methyl marks to prevent endonuclease binding.
She explained how the team at Johns Hopkins University have developed the hidden Markov model within nanopolish to establish the current distributions of methylated bases, including 5-methylcytosine (5mC), N6-methyladenosine (m6A) and 4-methylcytosine (4mC).
Utilising bacterial genomic DNA with known methylation motifs and the team observed some variability in signal shift; however, because each mark can be observed 6 times, it will be possible to reliably identify 5mC and 6mA. Detection of 4mC was also possible although the shift was weaker, which Yunfan, suggested may be due to a variety of biophysical properties, such as base stacking and changes in persistence length (the maximum length at which a polymer behaves as a stiff rod).
The team now plan to call modified bases in similarly methylated plasmids to validate their model.
Metagenomics Breakout: MinION microbiome profiling: going from on-the-go to go-to? –
Arwyn Edwards
Arwyn Edwards from Aberystwyth University spoke about using Oxford Nanopores Minion device for microbial profiling in the field. Highlighting the gaps in the earth microbiome project, Arwyn suggested that a decentralised sequencing approach could be what is needed to fill in areas with missing data.
Employing an “…offline off-grid sequencing protocol”, using battery powered DNA extraction, sequencing and bioinformatics workflows, Arwyn and the team first attempted to profile microbial communities at the bottom of a U.K. coal mine. One of the main issues was a battery powered centrifuge which, on one occasion, generated a spark which could have potentially caused an explosion by igniting the methane commonly found in coal mines. While initial attempts to sequence the microbial communities at the bottom of the mine were hampered by low DNA yields and the risk of death, further optimisation resulted in a successful glimpse into the microbial communities residing in this isolated hostile environment.
Using the method Arwyn and the team developed for off-grid sequencing they went “into the cold”. Here they attempted to profile microbial communities in cryoconite, windblown deposits comprised of rock particles and microbes which build up in the snow and ice of Artic areas resulting in microbial darkening. Shotgun metagenomics of samples from Greenland and Svalbard using the Minion system identified the expected “keystone” taxa showing the utility of their method. This enabled Arwyn to design hypothesis driven microbial biogeography studies in examining these systems.
Using a rapid 16S rRNA gene sequencing approach, Arwyn and his team examined the microbial communities of closed, snow coved cryconites and compared these with ones open to the air. Ordination methods and stacked bar charts were shown to visually describe microbial community dissimilarity at the family level, and that open and closed cryoconites exhibited a significant clustering within their experimental groups. Continuing with the microbial biogeography theme Arwyn then looked at the microbial community composition of Arctic and alpine cryoconites using rapid 16S barcoding showing that the two areas had distinct microbial communities at multiple taxonomic levels. Comparing these results with previously generated 16S rRNA gene amplicon pyrosequencing data on the same samples, proportional abundances of specific taxa and taxa ratios showed significant strong positive correlations between the two methods.
Coming “in from the cold” Arwyn re-sequenced an alpine cryoconite sample with Oxford Nanopores new field kit comprised of lyophilised reagents which had been stored at ambient temperature for 6 days. Comparing these results with those generated using the RAD kit and other short read sequencing methods, Arwyn showed that coherent microbial community profiles were consistently generated.

Mads Alberstsen from Aalborg University, Denmark
Mads spoke about the use of long read technology to aid in the assembly of microbial genomes from complex samples containing numerous different microorganisms. Mads described the two main limitations to genome recovery from metagenomes, these being micro diversity and the binning of the individual genomes. Micro diversity is the problem of having multiple closely related organisms in a sample, while binning the process of separating genomes into their constituent groups.
Using the analogy of re assembling a shredded newspaper, Mads spoke about the impact of micro-diversity upon assembly suggesting that it is more like reassembling a pile of newspapers. He stating that this isn’t a problem if the newspapers are different, but if they are the same it is very hard to assemble with just short reads.
In order to bin metagenomic data into discrete groups which represent individual genomes Mads suggested the use of differential coverage. This process uses the fact that the taxa abundance in a sample is maintained in the sequencing data and can be cross referenced over two or more samples. This results in distinct groupings of sequences representing data from discrete taxa. Furthermore as this approach is based on abundance, it is sequence composition independent. Showing a graph of this method applied to short read technology and Oxford Nanopores long read technology resulted in N50s of 5kb and > 100 kb respectively. One of the main pieces of advice that Mads offered to improve an assembly was to remove any shorter Nanopore reads in the data, ideally keeping anything over 4 kb. Subsequently Mads showed data to suggest that closely related organisms could be missed by short read technologies when used on their own, and that the inclusion of long reads dramatically helped detection. He finished by stating that long read data may provide a “[more complete] window into the microbial world”.
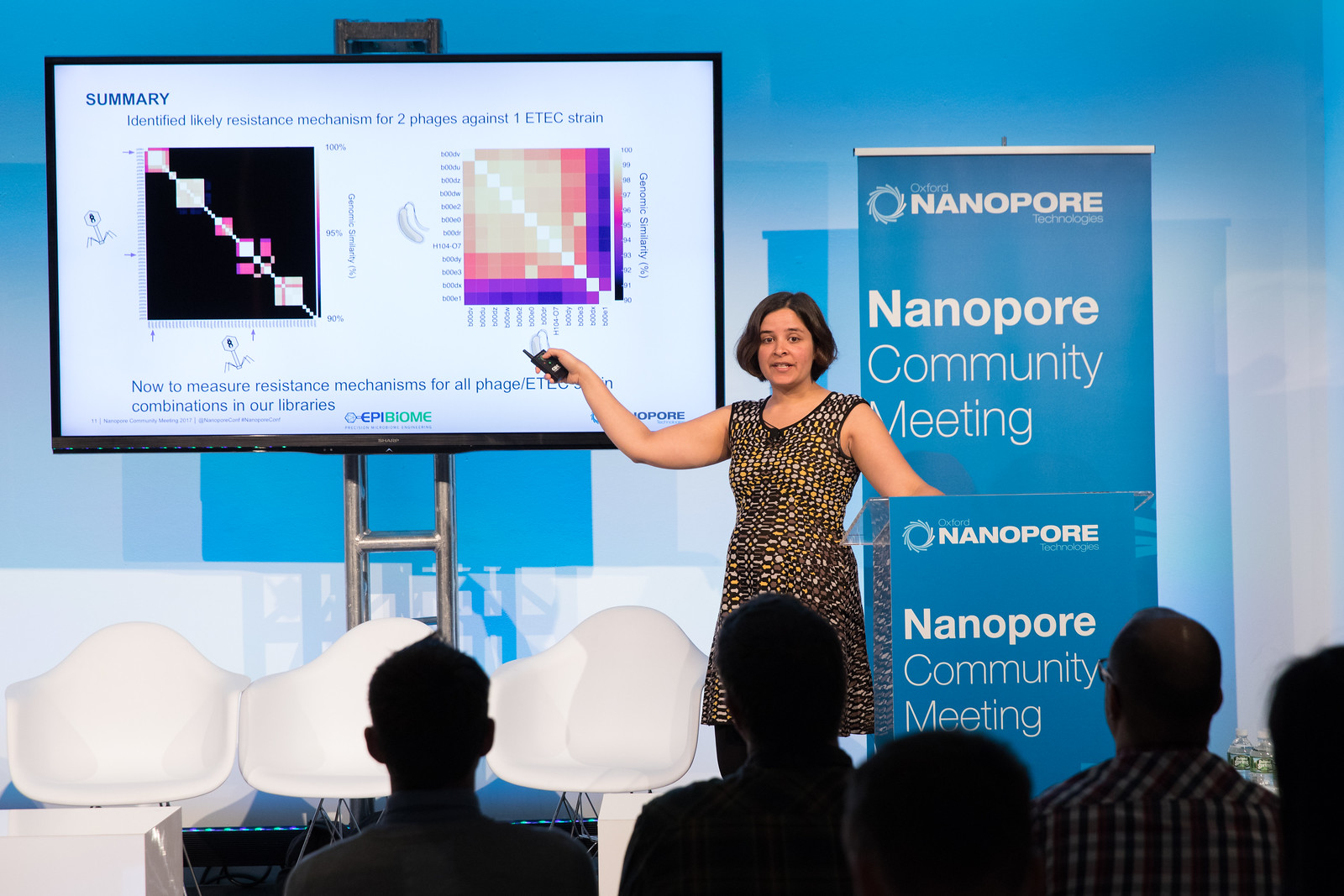
Understanding phage-bacterial host interactions for smarter therapeutics - Anika Kinkhabwala
Anika, from Epibiome, began her talk by highlighting the huge cost to human life antimicrobial resistance has. She stated that 700,000 deaths a year can be attributed to AMR and that this is projected to rise to 10 million by 2050.
Anika suggested that one potential way to combat antibiotic resistance is through the use of “the ultimate bacteria predator”, this being the bacteriophage. While highly specific to a bacterial host species or even strain, Anika and Epibiome are screening huge numbers of phage in a high throughput, automated manner. This is in the hope that they can create phage cocktails capable of fighting infections and thus reduce the reliance on antibiotics. Anika stated “It is our aim to find all the phages in the world!”
Understanding the genomic systems at work when phages and bacteria interact is crucial in determining synergistic or antagonistic behaviours they could exhibit. Using the Oxford Nanopore Minion device, Anika showed that a number of bacteriophage insensitive mutants (BIMS) had distinct genomic variation when compared with sensitive bacteria. It was theorised that these variations my result in changes to surface antigens giving the bacterium protection from specific phages which target these antigens. Knowing which phages are affected by which bacterial genomic variants helps in the design of optimal phage cocktails hopefully resulting in treatments with the broadest spectrum possible.
Jon Jerlstrom Hultqvist
Nanopore sequencing for genomics of heterotrophic protists
Jon Jerlström-Hultqvist from Dalhousie University began his talk with an overview of sequenced genomes describing a bias towards particular organisms. He noted there was distinct lack of whole lineages such as protists, and of those which have been sequenced, many were parasitic taxa. John stated he was particularly interested in free living protists as genomic information from these organism would give a good insight into their evolutionary history.
The main barrier for Jon was the fact that it is very difficult, if not impossible, to separate protists from prokaryotic organisms during culture for a number of reasons, one of the main ones being that often protists use prokaryotes as a food source.
One of the organisms that Jon was interested in was Meteora sporadica, an orphan lineage that is not closely related to any other eukaryotic super group. Using optimised culture, DNA extraction and bioinformatic methods, Jon was able to computationally split the sequences into prokaryote and protist bins. The Meteora sporadica assembly pipeline resulted in a prokaryotic assembly of 70 contigs and an N50 of 3.2 Mbp while the eukaryotic assembly consisted of 89 contigs of > 50 kbp and an N50 of 391 bp. Jon showed that this method produced a protist assembly with a BUSCO completeness score of > 95 % for his target organism.
PCE, an undescribed metamonad flagellate, was the second protist assembly attempted by Jon and his team. In this instance, it was known that less than 15 % of the 2 uG of genomic DNA extracted belonged to the target organism. Impressively the Eukaryotic proportion of this assembly had an average coverage of 55 X and comprised of 8 contigs all of which were above 50,000 bp. Jon stated that the PCE genome is now almost completely assembled and many of the contigs had telomeric sequences at both ends.
Jon finished by stating his take home messages that “…Protist genomics in complex communities [is] feasible with long reads” and “Long-reads give high quality assemblies, even at moderate sequencing depth”.
Breakout: data analysis/epigenetics
Matthew Riley
Matthew Riley of the US Army presented his research evaluating the MinION for biological defence applications. Whole genome sequencing was performed on a number of blinded bacterial isolates, which had previously been characterised using alternative methods. Two separate experiments were performed, where reference strain isolates were grown individually and then pooled together prior to sequencing. The data generated allowed identification of all strains down to the strain or species level within 2-3 hours of starting the sequencing run, Additionally, each species-level identification could be obtained using only 20% of the data captured. Matthew concluded that the MinION is currently capable of rapid identification of mixed cultured bacteria quickly, cheaply and with minimal infrastructure requirements. Furthermore, he suggested that the portability, low cost, small footprint and rapid turnaround makes nanopore sequencing an ideal candidate for field testing.
Wenqi Zeng
The team at Simcere diagnostics are developing methods utilising the MinION for a number of clinical and field applications. Wenqi Zeng from Simcere presented data from a recent study to identify culture-negative pathogens causal of endocarditis (an infection of the heart valve or inner lining of the heart). In all seven samples tested, nanopore sequencing allowed identification of the causative pathogen. Interestingly, the rare gram-negative organisms Coxiella burnetii and Bartonella quintana, were detected in two of the samples. The team have also used nanopore sequencing to successfully identify Dengue virus (DENV) from patient plasma, with one of 3 sequencing runs providing additional information on the presence of Acinetobacter baumannii co-infection.
Breakout: Larger genomes
Kim Judge – ‘Sanger sequencing’: Using GridION for large genomes in a core facility
Kim Judge, from the Sanger Institute, began by introducing the immense sequencing facilities at the Sanger – that to date have sequenced 4 petabases of data and are barrelling towards their 5th! The latest addition to their collection of sequencers is a GridION, and Kim continued by explaining why they have chosen to integrate the GridION into their production sequencing facility and the considerations that must be taken into account for effective management of the platform. In particular, Kim discussed in detail how standardisation of DNA quality assessment will be fundamental to providing consistent large-scale sequencing. For this, the Sanger Institute use FEMTO Pulse to examine fragment size, as it allows analysis of fragments up to 165 kb as well as working at a cost of roughly £4 per sample.
In addition to maintaining high quality standards, Kim went on to discuss how effective data management and storage must be planned and implemented before any large genome sequencing projects. She also described their new laboratory information management system (LIMS) – called Traction – for tracking the progress of samples they will sequence on their GridION through submission, quality assessment, library preparation and sequencing. Traction is available as an open source software tool for anyone that wishes to implement it.
Finally, Kim discussed plans for the Vertebrate Genome Project, in which the team at the Sanger aim to create reference quality, haplotype-phased assemblies of (eventually) representatives of all vertebrate species. The initial phase of the project will assemble the genomes of selected species to represent all the vertebrate orders. Kim explained the challenges of sequencing organisms with unknown genome sizes, and described how the Sanger Institute plans to employ a much more consultative approach when it comes to nanopore sequencing to obtain the best results – finding the compromise between throughput, read length and input DNA quantity dependent on the biological question being asked.
To conclude, Kim summarised that her team’s aims going forward were to investigate extraction processes for such a wide variety of samples, work on improving throughput per flow cell, and find methods to clean up precious samples without losing too much DNA.

Taco Jesse – Exploring new horizons in plant breeding using Nanopore sequencing
Taco Jesse, of Rijk Zwaan BV, began by describing the company and the value of plantbreeding and the seed market – for example, one kilo of tomato seeds is more expensive that one kilo of gold. Rijk Zwaan focus on quality, and with a world market value of ~3.5 billion euros and sales in over 100 countries they are exploring new technologies to achieve the best results. Rapid and cost-effective introduction of traits allows competitive performance in the vegetable seed market, so Rijk Zwaan BV have begun using nanopore sequencing to allow them to do this.
In addition, Taco celebrated the thriving Nanopore Community in the Netherlands – with four PromethIONs installed and hoping for a fifth at Rijk Zwaan! The R&D team there comprises experts in seed techonology, cell biology, biochemistry, phytopathology and many more specialisms.
Taco went on the explain their preliminary experiments and the need for long reads to resolve complex plant genomes including large structural variation and extensive retrotransposons. Starting with the melon genome, the team at Rijk Zwaan used 7 flow cells to generate 70x coverage, assembling the data with minimap2, miniasm, followed by visualisation with Bandage. The first draft de novo assembly took just one day to generate from the reads, and gave a fantastic result – unpolished data returned an N50 of 10 Mb with 12 contigs covering half of the genome. The team also discovered a TTTAGG repeat motif that would have been undiscoverable with short reads.
To finish, Taco presented their most recent results on an assembly of the tomato genome, from just last month. The total output for this project so far has been 45 Gb of data with 1D ligation libraries, and they are pushing to get an average fragment size of 30 kb regularly.
Their current focus is to optimise extraction methods to enable generation of the most ultra-high-molecular-weight-DNA possible – mainly in order to make the bioinformatics pipeline both easier and faster for assemblies of very large plant genomes.
Future work by the team will be focussed on exploring even more plant species, increasing the genome size as they go. Taco concluded by crediting Baptiste Mayjonade for his exceptional work on extraction protocol generation for plant species, and encouraged others to use these protocols in future.
Todd Michael – High molecular weight DNA enables ultra-long reads for genome assembly
Todd Michael, from the J Craig Venter Institute, begun by reiterating the theme of the breakout so far – that very high molecular weight DNA and correspondingly long reads make for the easiest and quickest assemblies of larger genomes. Todd explained how his team always look for the “smile” in gel electrophoresis of the sample to indicate the presence of high molecular weight DNA.
Todd went on to outline some of the difficulties associated with genome assembly, particularly that of larger eukaryotic genomes. More specifically, resolution of organelle genomes and complex regions such as centromeres or nested transposable elements, remain a considerable challenge. To address this problem, Todd and his colleagues would like to generate the longest reads possible, capable of spanning some of these more difficult stretches and enabling their assembly. To achieve this, as little shearing as possible introduced into the process to maintain lengths of DNA for long read sequencing. The most recent technique with which they have seen success is in-gel extraction without shearing. Following HMW gDNA extraction, Todd uses 1D library preparation.
As an example of how these long reads can be beneficial, Todd used an example of their work sequencing Arabidopsis thaliana – a well characterised genome of ~115 Mb but still with one significant question unanswered about the biology of the genome. This question is: if you insert genes into the Arabidopsis genome, what do these insertions look like? Agrobacterium inserts DNA at random, but these are long inserts and the architecture is not known, as the technology did not exist to determine it.
Assembling an Arabidopsis sample took the team just 4 days from sample collection to finished assembly using minimap2 and miniasm and gave full chromosome-sized contigs. The only sections that could not be assembled were the centromeres – which are stretches of identical 178 bp repeats that run for megabases of length. Todd explained that he has reads covering this area – but they simply cannot be assembled. He went on to reiterate that even with a small genome, highly repetitive cassettes have a big impact, and in fact using their assembler of choice (Canu), the assembly took over a week.
As a second example, Todd described how he began sequencing giant kelp, which is an important indicator of ocean health, and also gets in the way when you are surfing in California! One flow cell gave 10x coverage of the nuclear genome, 500x coverage of the chloroplast genome and 50x coverage of the mitochondrial genome. The mitochondrial genome is 37 kb, and so there were multiple reads in the dataset that spanned this complete length. This allowed identification of heteroplasmy between mitochondria, high variability and at least two major haplotypes – indicating that in a large plant there are lots of mitochondria that are often changing. This show potential for nanopore sequencing to enable a different way of looking at organelle genomes.
Todd concluded by referencing the things he had learnt at the conference so far, particularly the existence of the whale-watching competition for ultra-long reads, to which he may have some hidden gems to contribute!
Plenary:
Nanopore cDNA sequencing produces complex transcriptomes for sensitive and accurate gene expression profiling - Eshita Sharma
Eshita, from Oxford Genomics and the Wellcome Centre for Human Genetics, began by describing thecore facility and the applications they provide. To ensure they provide a good service to customers, Eshita said that they aim to offer the most up-to-date technologies possible, and are now looking to provide Oxford Nanopore as part of this portfolio. As over 58% of the requested services involve polyA RNA sequencing, Eshita was interested in comparing Oxford Nanopores PCR cDNA kit with their current short read services. Some of the main reasons behind the desire to evaluate the cDNA kit was that the long reads provided by Nanopore technology are perfect for isoform detection.
Eshita and the team decided to evaluate the PCR cDNA kit using a number of spike-in and RNA control molecules. These were the Universal Human RNA Reference (UHRR), External RNA Control Consortium (ERCC) and spike-in RNA variants (SIRVs). These samples were run using both Oxford Nanopore's PCR cDNA kit and the current short read service pipeline they currently provide.
Using the Oxford Nanopore PCR cDNA protocol to sequence these samples resulted in 2.2 Gb and 3.1 Gb of mapped sequences over two replicate runs, and this provided 50% and 51% of the transcriptome respectively. A short-read dataset starting with 15.2 Gb gave 77.1% of the transcriptome.
When analysing the ERCC spike in controls Eshita said that both methodologies show comparable numbers of detected spike-ins and that spike in dropout rates was similar when subsampling was implemented. The linear dynamic range of both methods was approximately 215 and there was a high correlation in signal abundance plots (R2> 0.94). When examining fold change estimates using the ERCC spike in controls, both methods produced similar results. Similarly with the UHRR data both technologies showed comparable numbers of detected genes and again dropout rate was similar when subsampling was implemented.
Eshita went on to describe the expression profiles for the UHRR sample and how they correlated between the two methods. Eshita quotes a correlation coefficient of 0.7 and noted that they saw several genes with high counts in the Nanopore data which were missing or low in the alternative technology. When interrogating the data further Eshita saw that these genes were shorter in length.
In conclusion Eshita said that they got high quality complex transcriptomes from Nanopore cDNA sequencing and that their current technology has a similar dynamic range; limit of signal detection; and fold change responses. Furthermore transcriptome complexity was comparable in terms of detected genes and coverage at lower sequencing depths.
Plenary: Charles Chiu
Dr Charles Chiu from UC, San Francisco, closed the meeting by summarising his ground-breaking work implementing metagenomic sequencing in the clinic. He outlined his involvement in the Precision Diagnosis of Acute Infectious Disease (PDAID) study. This multi-hospital study of 215 patients, aims to assess the utility, outcome impact and cost-effectiveness of CLIA-validated metagenomic testing versus conventional microbiological testing (protein testing, antigen testing, PCR, RT-PCR and culture) for the identification of meningitis and encephalitis causing pathogens. neurological infection was identified in 22% of cases. This may seem a low number; however, to enrol in the study, patients had to have had no diagnosis from conventional testing.
A significant advantage of metagenomic testing, is that is it hypothesis-free, which means clinicians don’t have to have a preconceived idea of the causative agent. This is particularly important for a range of infectious diseases which present with the same or similar symptoms, confounding conventional diagnosis. Charles gave the example of acute febrile illness, which can be caused by a range of bacteria, viruses and other organisms. Such infections are often treated empirically with a range of antimicrobials, which increases healthcare costs and is not good medical practice. A single metagenomic analysis, rather than sequential testing with traditional techniques clearly holds much promise for timely detection and therapeutic intervention.
Continuing the theme of rapid detection, Charles also highlighted how sepsis (where the body’s own response to infection causes damage to tissues and organs) has a mortality rate of approximately 50% if the appropriate antibiotic is not administered within 4 hours of admission to a healthcare setting. With this startling statistic in mind, Charles presented data generated in his lab, showing how, using the real-time analysis capabilities of the MinION, was possible to detect Ebola virus (EBOV) and chikungunya virus (CHIKV) within just 6-8 minutes of a sequencing run — with a total time from sample to result of approximately 3 hours. Importantly, the shotgun metagenomic sequencing approach used was not targeting a specific microorganism, rather it was sequencing everything in the sample. Further, Charles described that how, using a ‘spiked primer’ approach, his team has been able to achieve rapid and highly sensitive detections of pathogens from clinical body fluids down to just 102 copies per ml. Importantly, this approach highly sensitive detection without impacting breadth of metagenomic detection.
In order to streamline their metagenomic analyses, the team at UCSF has developed ‘SURPIrt’, a real-time computational platform that can analyse up to 1 million reads in <10 minutes on a laptop. Charles stated that this compares very favourably with the both the infrastructure and time requirements of traditional short-read sequencing platforms. They have developed an entirely portable, field-ready assay, allowing real-time detection of infectious disease in remote or limited infrastructure environments. Using this workflow, they can detect a whole range of organisms from fungi, to bacteria and viruses in within 2 minutes or 2 hours of starting a sequencing run for very low titres.
Charles closes his talk and the conference by describing his team’s current work to clinically validate the nanopore platform in a CLIA lab.






