Nanopore Community Meeting 2017: Day 1 write-up
Day 1 of the Nanopore Community Meeting is done, and if you were not there in person, you may have been following #nanoporeconf on twitter.
Videos of the sessions will be released soon. In the meantime, you can find some brief summaries of the sessions below.
Plenary: Nick Loman, University of Birmingham
Nick Loman, Professor of Microbial Genomics and Bioinformatics at the Institute for Microbiology and Infection at the University of Birmingham kicked off proceedings with his concept of ‘sequencing singularity’, where a whole raft of diagnostic tests could, in the future, be replaced by a single sequencing assay. Such a test would allow pathogen detection, phenotypic profiling, source attribution, outbreak tracking and host response to infection and treatment.
Nick reviewed the recent advances in nanopore sequencing, including his team’s collaborations on viral and bacterial diagnostic sequencing, real-time surveillance, direct RNA and human whole-genome sequencing.
Nick reviewed his team’s role in tracking the Ebola epidemic in West Africa between 2013-2016. The MinION project in Guinea in 2015 provided rapid insight into viral transmission, allowing enhanced outbreak management. The MinION could be transported in a small suitcase and deployed in the field, delivering real-time results within a couple of days. Using the nanopore data, the team uncovered that Ebola was frequently moving long distances within and between countries. Furthermore, they identified that new Ebola flare-ups were transmitted via previously infected survivors, rather than from animal reservoirs as had been suggested. In one example, Ebola virus was transmitted via a survivor 500 days after initial viral infection, resulting in a new ‘flare-up’.
Nick opined that in the future, such portable, real-time sequencing could prevent outbreaks becoming epidemics. In order to further develop the infrastructure to make this belief a reality, the team applied the finding from their studies of Ebola to the Zika outbreak in Brazil. Unlike Ebola, Zika has very low titres of virus in clinical samples and, as result, standard metagenomic sequencing does not work.
To address this issue, the team developed a new tiling PCR approach, which permitted the genome to be sequenced, which in turn allowed more effective tracking of Zika spread across the Americas. The data revealed that the first clinical diagnosis of Zika infection was made over a year after the virus became prevalent in the region – highlighting the limitations of existing surveillance techniques.
Nick then turned his attention to the application of sequencing to pathogen detection in the clinic. While he believes that sequencing is poised to disrupt clinical practice, he pointed out a number of challenges that still need to be addressed to make this happen. Amongst other things, these challenges include simplifying informatics and wet-lab workflows. His team are addressing these issues, with Nick highlighting some recent work on rapid library prep protocols that have reduced their sample prep time in half, to approximately 4 hours.
Another challenge Nick’s team is working on is ensuring the assay works for a wide variety of pathogens, while overcoming the issues such as the high levels of host DNA found in most clinical sample types.
Closing his presentation, Nick congratulated Martin Smith from the Garvan Institute for Medical Research for the longest nanopore read, of 970kb. However, he intends to hold on to his team’s record of sequencing an entire E. coligenome using 8 ultra-long reads, with an assembly time of just 1.5 seconds. Based on which, Nick posed the question: “are we now looking at the end of genome assembly?”.
Nick was interviewed after his session:
Plenary: Sissel Juul, Oxford Nanopore. Applications in genetics and genomics using nanopore DNA, cDNA, and direct RNA sequencing.
Dr. Sissel Juul, Director of Genomic Applications at Oxford Nanopore Technologies opened her presentation by showcasing the Applications group scientific posters which are available online here. Of note were posters describing:
· Advances in the VolTRAX system, an automated preparation device currently with early users. Version 2 in 2018 will include PCR and DNA quantification and a WGA from a few cells by on device library prep;
· Comparing de novo assembly of genomes with 1D and 1D2 sequencing showing that 1D2 produced a more contiguous assemblies than 1D and this effect was most notable when shorter libraries were analysed.
· Using long Nanopore reads to assemble bacterial genomes from complex samples such as soil. This showed that even when full assemblies were not achievable, the methodology provided valuable taxonomic and functional insight into complex biological systems.
Sissel introduced the group’s research into detecting abnormalities in chromosome numbers and a potential increased risk of miscarriage, through pre-implantation genetic screening (PGS) using the Oxford Nanopore MinION system. In collaboration with Simon Fishel of CARE Fertility, Sissel described how whole genome amplification upstream of nanopore sequencing could be used to detect aneuploidy. Concurrent targeted PCR could also detect the ANXA5 M2 haplotype, a genetic marker known to increase the likelihood of recurrent miscarriage. By screening a blastocyst biopsy soon after egg collection, time and cost of the whole procedure could be dramatically reduced, while control of the process could be maintained. Furthermore, both assays can be performed simultaneously by using the WGA amplified material as an input for a low PCR cycle assay amplifying the ANXA5 M2 target. In sensitivity tests, aneuploidy was robustly detectable at the low coverage of approximately 25 Mb. At a sequencing speed of 450 bp per second this allows the generation of results in less than 5 minutes of sequencing time on a MinION.

Structural variation in cancer was the next topic and Sissel showed that extensive inter- and intra- chromosomal rearrangements were revealed specifically via the use of the long-read nature of Nanopore sequencing. At between 15 and 19.5 X coverage of two cancer cell lines, known non-synonymous mutations in the KRAS gene were clearly visible in the sequencing data. These results showed that long nanopore sequence reads could be successfully used for the analysis of tumour genomic DNA, and for the identification of SVs with far higher resolution than karyotyping
On RNA sequencing, Sissel reviewed the three new kits released this year by Oxford Nanopore. Sissel showed that Direct RNA sequencing, PCR cDNA sequencing and PCR free cDNA sequencing had negligible GC content bias or read length bias. One poster showed that 50-fold fewer reads and 7 fold fewer bases were needed to detect the same number of fully covered (> 95 %) transcripts, than short read technologies. The PCR-free cDNA method was then used to perform comparative transcriptomics of E.coli under different growth conditions and highlight differentially expressed gene sets between the two.
Long non-coding RNAs (lncRNA) were also discussed and a specific set of poorly characterised transcripts, thought to have a cancer killing phenotype, were investigated. As only a small conserved region of the transcript family was known, a semi specific PCR was used to identify the 5’ and 3’ ends of the transcripts and numerous complete, and never before seen, isoforms were found.
In the final section Sissel went on to talk about new methods developed by Oxford Nanopore to detect modified nucleotides in both DNA and RNA. Using the newly released Tombo modified base detection algorithm, a secondary signal level shift was detected when basecalled genomic data containing modified bases was analysed using the new model.
In Sissel's concluding remarks she discussed the current and future scope of applications available to MinION users. These included genomic applications such as; forensics, water testing, consumer genetics, biodefence, authentication, food safety and many more.
Sissel was interviewed after her plenary:
RNA/cDNA Breakout:
Transcriptional Landscapes analysis through direct RNA sequencing - Intawat Nookaew
Prof. Intawat Nookaew from the Dept. of Biomedical Informatics at the University of Arkansas spoke about overcoming problems associated with sequencing repeat regions in genomic DNA by using Oxford Nanopore’s long read technology.
Intawat then went on to speak about using direct RNA sequencing to determine gene expression patterns as yeast switch from glucose to ethanol as a growth substrate. Replicate samples under the different growth conditions and phases showed distinctive gene expression profiles, clustering within their respective experimental groups on an ordination plot. This elegantly demonstrated how standard differential gene expression methods could be applied to ONT direct RNA data. Furthermore, gene-set analysis using Piano highlighted specific genes strongly associated with each experimental group. Organisms using ethanol as a growth substrate had significant fold increases of genes related to ethanol metabolism, while up regulated genes in the glucose group were associated with rapid growth processes.
Comparing different RNA-seq methods, 0.5 Gb of direct RNA sequences gave comparable results to 1 Gb of reads generated by alternative short read technologies. Furthermore there was evidence to suggest that the longer nanopore reads had a lower GC bias.
Intawat finished his talk by discussing ways to uncover transcriptional structure in molecules such as premature mRNA, dual transcripts and non-coding RNA.
Direct RNA sequencing of Influenza viral RNA using the MinION nanopore sequencer - Matt Keller
Matt Keller, from the University of Georgia, spoke about direct RNA sequencing of seasonal influenza A. He started by describing the health impact this virus has as it can cause up to 1 million hospitalisations a year. As this organisms has a segmented negative sense RNA genome Matt suggested that this made it a perfect system for investigation by direct RNA sequencing on the nanopore platform.
In order to attach direct RNA sequencing adapters to the viral RNA, Matt used a method that exploited two conserved regions of the genome and designed adapters which were ligated to these areas.
Virons cultured in egg allantoic fluid were purified by two separate methods in order to be used as a template for direct RNA sequencing on the Oxford Nanopore platform and short read cDNA sequencing on the MiSeq platform. The first purification method was a long ultracentrifugation protocol which resulted in a very pure sample while the latter was a quick centrifugation method which produced a crude sample cleared of cellular debris.
Matt stated that while the short reads performed well, producing (cDNA) reads which had low average error rates, the nanopore direct RNA data produced reads spanning the entire viral genome and sequenced the actual molecule of interest rather than a synthesised copy. While the average read error rates were comparably high, Matt spoke of technology improvements that could mitigate some of these issues.
Both the pure and crude sample prep methods produced RNA sequences with consensus accuracies around of 99 % and 98.7 % respectively and furthermore the percentage of direct RNA reads mapping to the influenza genome were over 95 %. Matt finished his talk by discussing some of the potential uses for direct RNA sequencing in this context, including the detection of modified bases.
RNA sequencing and data analysis using the MinION system - Stéphane Le Crom
Prof. Stéphane Le Crom from Pierre et Marie Curie University Paris is the scientific head of the IBENS genomic core facility and provides sequencing services for researchers working on high throughput genomics, specifically functional genomics based projects.
Providing end-to-end support, from experimental design to data analysis, Stéphane and his team developed a number of software packages to aid in this. ToulligQC, a package for RNA sequencing evaluation and barcode demultiplexing, provides QC metrics for nanopore RNA sequencing runs and was used by Stephane and his team to determine that barcodes had no visible influence on the overall throughput of sequencing.
With the introduction of a new technology, Stéphane and his team validate new methods using a knock out mouse model with blocked myelination. Gene expression profiles are then compared against the wild type phenotype. Using this model system, MinION cDNA data and NextSeq cDNA reads were aligned to the mouse reference transcriptome by Minimap2. The two datasets showed a good correlation and of the 6551 differentially expressed genes detected in the nanopore data, 86% were shared with the NextSeq dataset. Most interestingly new and novel transcript isoforms for the TMP3 gene were detected using the nanopore cDNA sequencing methods and were not detected using the short read methods. Furthermore, it was shown that low read numbers (100k reads) could be used successfully to detect differentially expressed genes.
Stéphane and his team were early users of the new PCR cDNA kit and quickly broke their own read records with 7.5 million reads from a yeast sample and nearly 10 million for an ERCC spike in control mix. With use on their own “real” samples, consistent high read numbers were also produced. Using this new technology, and their own in house developed software, IBENS are aiming to provide a nanopore-specific RNA Sequencing pipeline for end-to-end differential gene expression analysis.
‘Squiggle'-cell sequencing - Martin Smith
Martin Smith from the Garvan Institute spoke about single cell sequencing methods using Oxford Nanopore's long read technology. Martin began by explaining the methodology used to perform single cell sequencing using the Chromium 10X platform. Here single cells are separated mechanically and specific barcodes can be added during cDNA generation. This is done in order to identify which transcripts came from which cells when cDNA from many cells are sequenced at the same time.
In order to perform single cell transcriptomics it is important to be able to demultiplex the barcodes accurately. Martin then went on to state “… [There is] no inherent error in the underlying [raw] data…” and suggested that accurate barcode detection could be performed in “event” or “squiggle” space if flanking sequences are known in “base” space. Martin showed that the polyA sequence and PCR primer sequences in the cDNA molecules could be found in the basecalled data and then, by knowing the location of these regions in the underlying event data, extract the single cell barcodes in event form. Using an unsupervised clustering method the extracted barcode events could be grouped together and thus the cell of origin could be determined.
Martin then spoke about using a similar method that could be used to identify the standard Nanopore barcodes. Although it did not perform as well as Albacore, Martin suggested that it would be a useful approach to screen the “unclassified” barcodes rejected by Albacore in order to rescue those with a good quality score.
When comparing a basic text search for barcodes against his “squiggle cell sequencing” method of barcode detection, Martin found that his approach identified barcodes at a much higher rate.

Completing genomes breakout:
Ryan Wick
Ryan Wick, from the University of Melbourne, focused his talk on completing microbial genome assemblies. Ryan initially described how assembly is sometimes thought of as a “solved problem” – but while longer reads help to overcome some of the issues associated with assembling short read data, there are still significant improvements to be made.
With the goal of achieving the perfect assembler, Ryan outlined the ways in which assembly can currently fail, examining the roots causes and possible solutions to each.
Using an example of a Klebsiella assembly, Ryan explained the different stages of assembly that lead to different levels of completeness. Initial fastq assembly returned an accuracy of 99.5%, and use of nanopolish raised this to 99.7%. Methylation-aware nanopolish increased this number to 99.9%.
Ryan continued by describing how short read polishing can help to fix a lot of errors- but crucially only in non-repeat regions. Giving several examples, Ryan showed how repeat regions can significantly hinder assemblies, generating failure in various circumstances such as stretches of tandem repeats where the number of repeats cannot be identified.
Further than this, though, there are several situations in which assemblies fail for a reason that isn’t clear, and we need to work out where the shortcomings in assemblers are, as some datasets can assemble in one assembler but not another. In some situations, Ryan explained, the assembler needs to make a decision, but this decision is not clear cut. For example, what if there is variation within an isolate sample? What if your sample has two plasmids that are 70% identical? Should the assembler go with majority rule, separate the assemblies or graph the variation?
Ryan finished by discussing how we always need longer reads and more intelligent assemblers – as these problems are applicable across multiple scenarios, for example cancer samples with very high heterogeneity. These problems, though, are being actively worked on by a lot of brilliant researchers – so we will continue to see things change.
Concluding, the audience are reminded that Ryan will demonstrate his bacterial genome assembly pipeline, comprising of Albacore, Porechop, Unicycler, Filtlong, Bandage in the data analysis tools session over lunch.
Conrad Stack
Conrad Stack, from Mars Inc, completed the breakout by discussing the application of nanopore sequencing to create reference genomes for plant species. In this validation of the technology, Conrad and his team sequenced the ‘Pound 7’ clone of cacao that is heterozygous and estimated to be 442 Mb in size, generating a high-quality reference genome.
The aim of Conrad’s team’s work was primarily to the characterise the genomic diversity in the 10 primary ancestry groups of T cacao and related Theobroma species, alongside additional supplementary aims of building a high-quality reference genome, comparing different levels of assembly polishing/finishing, and validating resolution of the complex heterozygous QTL locus.
Conrad described previous assembly efforts using BAC sequencing that took over two years to complete – showing diagrams of active transposable elements regions with complex structural variation. Comparing these BAC-derived haplotypes to the MinION contigs demonstrated that the nanopore sequencing was able to capture this incredibly complex repeat region almost completely. Conrad also outlined how addition of HiC or other scaffolding data might help to continue to resolve high-complexity regions typical of plant genomes.
At an estimated cost of less than $5000, this reference was compared to several previous reference assemblies from 2010, 2013 and 2017. The first assembly from 2010 had over 25,000 contigs, but Conrad’s team’s most recent efforts resulted in just over 1,000 contigs.
In conclusion, Conrad summarised the good, the bad and the ugly – challenges remain in terms of generating high coverage of plant genomes and polishing data for the best possible assembly, but at a low cost with the ability to resolve complex structural variation and generate single contigs of organelle genomes with no additional effort, nanopore sequencing could be influential in progressing genome-guided plant breeding.
Structural variations breakout:
Chia-Lin Wei
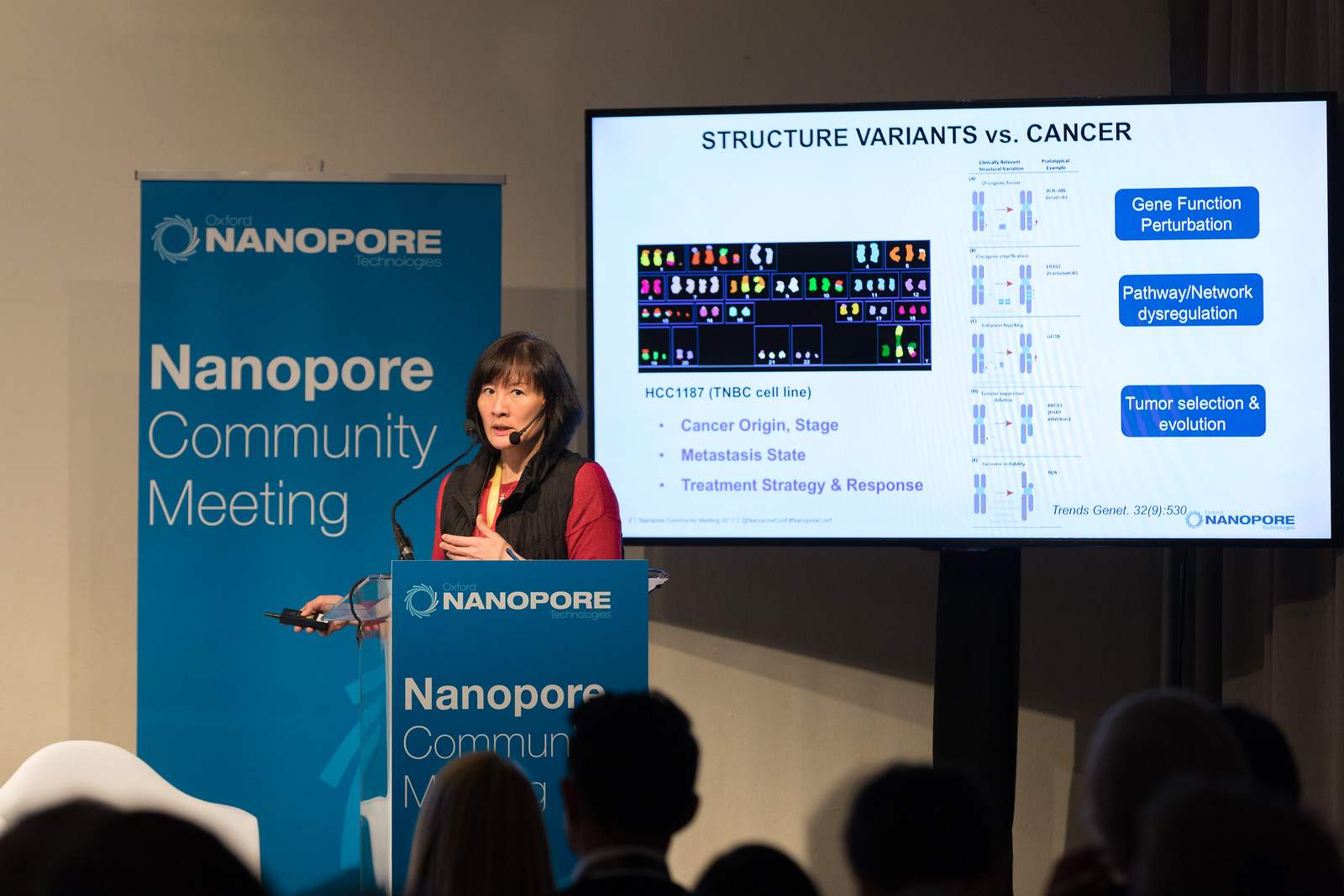
Chia-Lin Wei from the Jackson Laboratory discussed her work utilising nanopore sequencing to identify structural variation in the cancer genome. Structural variants (SVs) are common in the cancer genome; however, the short-read lengths delivered by traditional sequencing technologies makes the analysis of such genomic rearrangements extremely challenging – obtaining read lengths of 10 – 50 kb significantly reduces the depth of coverage required. Chia-Lin and her team utilised the long-reads provided by nanopore sequencing in combination with a customised analysis pipeline, Picky, to reveal significant and diverse SVs in a breast cancer model including tandem duplications, deletions and translocations. Using PCR across the breakpoints, SV calls selected with two reads support were 100% validated. Using modest sequencing coverage, Chia-Lin stated that the team: ‘identified the full spectrum of SVs with superior specificity and sensitivity relative to short-read analyses and uncovered repetitive DNA as the major source of variation’.
Furthermore, nucleotide-resolution analysis revealed micro-insertions as a common feature of SV breakpoint. Breakpoint density across the genome is associated with propensity for inter-chromosomal connectivity and transcriptional regulation. Chia-Lin commented that the ‘utilisation of real-time, long-read sequencing to characterise SV in cancer cohorts will facilitate strategies to monitor genome stability during tumour evolution and improve therapeutic intervention’.
Joseph Schacherer
Joseph Schacherer, Professor of Genetics and Genomics at the University of Strasbourg, described how comprehensive genomic variant maps are essential to explore genome evolution and its phenotypic consequences. In humans, a large part of phenotypic variance is unexplained by characterised mutation types. This ‘missing variation’, may, in part be related to the traditional short-read sequencing technology used for many genome sequencing projects, which is unable to provide accurate characterisation of structural variations, (i.e. indels, inversions and translocations).
In order to overcome these challenges, his team are now utilising the MinION, which delivers ultra-long reads capable of spanning large structural variations (SV) and repetitive regions. Joseph told the audience how, at 20x mean coverage, the MinION delivered highly complete and contiguous genome assemblies of the yeasts Saccharomyces cerevisiae and Dekkera bruxellensis. Yeasts are powerful model systems to understand population genomics as they have broad genetic and phenotypic diversity. In their hands, SMARTdenovo proved to be the most effective assembler; however, Joseph recommended researchers try a number, to see which suits their experimental conditions. The high-quality assemblies generated enabled accurate detection and characterisation of SVs. Furthermore, the team were able to provide a complete cartography of transposable elements for both genomes.
The team now plan to apply the lessons learnt in this initial research to larger population studies. In summary, Joseph commented that: “preliminary results clearly show the value of the MinION system for screening whole genomes for complex SVs and deep characterisation of genome architecture in natural yeast populations”.
Svetlana Madjunkova
Svetlana Madjunkova, from the CReATe Fertility Center, shared her recent work evaluating the potential applicability of nanopore sequencing for pre-implantation genetic diagnosis (PGD) - a technique whereby in vitro fertilised embryos are tested for potential deleterious chromosomal rearrangements, allowing the implantation of euploid embryos. The technique is commonly employed for known carriers of chromosomal rearrangements, who are at higher risk of reproductive failure.
Svetlana opened her presentation by stating that we are in a brave new world of reproductive health, as new techniques such as pre-implantation genetic screening (PGS) and PGD are being driven via the advent of high-throughput genetic analysis tools, such as aCGH and NGS. However, there is still progress to be made, as these tools are limited in their facility to detect carriers of balanced translocations. While short-read sequencing can detect balanced translocations, the time, cost and resource requirements make it prohibitive. According to Svetlana, the ultra-long reads delivered by nanopore sequencing make it attractive technique for PGD.
The team at CReATe established a protocol and bioinformatic pipeline for PGD utilising the MinION which enabled accurate breakpoint mapping of balanced translocations in the carrier partners of 5 couples. Based on the data, custom breakpoint PCR assays were developed for each translocation, allowing high-sensitivity testing of the 21 embryos produced by the couples.
In closing her presentation, Svetlana concluded that: ‘Long-read MinION sequencing is an emerging solution for high resolution comprehensive clinical preimplantation genetic diagnosis of chromosomal rearrangements including balanced carriers’.

Lightning talks
Rachel Rubinstein
In our first Lightning talk for the meeting, Rachel Rubinstein from Ginkgo Bioworks presented her work utilising the MinION and What’s In My Pot (WIMP) workflow to enable rapid identification of bacterial contamination in an industrial fermenter. She described how existing methods such as traditional short-read sequencing technologies are too time consuming for the requirements of an industrial fermentation setting. In stark contrast, real-time nanopore sequencing allowed the identification of a bacterial contaminant in a fermentation run within 8 hours from receipt of sample. This rapid turnaround allowed decisive action to be taken, protecting the fermentation product and, in doing so, saving the organisation approximately $100,000. Rachel suggests that in future, such technology could be used for real-time monitoring of fermentation.

Jeffrey Rosenfeld
Jeffrey Rosenfeld from Rutgers Cancer Institute of New Jersey, presented data on the use of long sequencing reads to characterise tumour DNA. In breast cancer, the HER2 amplicon has an important if not yet fully understood role. The copy number of genes in the HER2 amplicon are known to increase but the exact organisation of these duplicated and potentially rearranged genes is difficult to characterise using traditional short-read approaches. Jeffrey presented data showing how nanopore sequencing allowed this gene structure to be determined, although some gaps remain, which the team plan to close. The gaps seem to line up with with high and low GC regions, so the goal is now to try different capture strategies or low-coverage whole genome sequencing. Jeffrey states: 'As throughput increases, the benefit of capture or targeted sequencing is outweighed by its cost'.
Sebastiaan Theuns
Sebastiaan Theuns from Ghent University revealed how nanopore sequencing is providing new insights into viral enteric disease complexes for improved animal health management. He explained how novel viruses are being increasingly discovered in the faeces of piglets, emphasising the need for improved diagnostic surveillance and tools. Using a metagenomic approach on unamplified DNA, the team detected a kobovirus, which had not previously been seen in Belgian livestock. Using the MinION, they were able to identify viral reads within just 7 seconds of the sequencing run. Sebastiaan reported that further surveillance of the affected farm is in progress and described the MinION as a useful tool for diagnosis of enteric disease in pig medicine. In conclusion, he stated ‘We are strongly convinced that this technology will be disruptive to the whole diagnostic market’.

Sarah Stahl
Following on from previously released data, where the MinION was successfully used on board the International Space Station (ISS), Sarah Stahl from NASA Johnson Space Center provided an update on the first ever complete sequencing workflow – from sample to result – to be undertaken in space. In a proof-of-principle study, a microbial mock community was sequenced with all sample processing steps, including DNA amplification and library preparation, carried out on board the ISS. Following the successful outcome of this experiment, the team went a step further, performing 16S sequencing of microbial organisms collected from and cultured on the ISS. Using the MinION, microorganisms could be rapidly identified at species level, demonstrating how this workflow could be used for environmental monitoring of space missions. Analysis of the returned samples using biochemical methods and Sanger sequencing gave concordant results to those obtained on the ISS. Sarah commented that: ‘This was a huge milestone for microbiology and we are very excited to continue using nanopore in the future for research on the ISS and beyond. The ability to prepare and sequence DNA in the spaceflight environment will revolutionise space-based research and in-flight medical operations’.
Chang Liu
Chang Liu from Washington University At St Louis presented his research utilising nanopore sequencing for high-resolution typing of human leukocyte antigen (HLA) genes. The HLA genes are the most polymorphic in the human genome and the accurate typing of these genes is critically important to the successful, long-term outcome of organ or stem cell transplantation. Chang previously presented data showing that nanopore 2D reads allowed highly accurate HLA typing with just 50 reads. In this talk, he demonstrated the feasibility of using 1D reads for HLA typing. He further explained how these long reads deliver direct allele phasing, something that is not easily achievable with short-read sequencing technologies. Chang described his approach of multiplex PCR, targeting full genes or key exons of a number of HLA loci, combined with barcoding to deliver further cost savings. According to Chang, the results indicated that nanopore sequencing is a promising technique for HLA typing.
Devin Drown
In the final Lightning talk in this session, Devin Drown from the University of Alaska Fairbanks presented his latest research applying nanopore sequencing to reveal natural antibiotic resistance in microbes associated with thawing permafrost. Alarmingly, Devin stated that, for the past 50 years, the artic has been warming twice as rapidly as the world as a whole. As climate change thaws the permafrost, ancient microbes with natural antibiotic resistance may be released with potential negative impact on human and animal health. Previous studies had identified that natural antibiotic resistance is found in ancient microbes within the permafrost, so the team wanted to investigate how common this resistance is in boreal forest soil microbial communities. It turns out, resistance is wide spread. Using metagenomic analysis, the team identified individual resistance genes which will enable improved understanding of the environmental reservoir of antibiotic resistance in Alaska.

Plenary: Steven Salzberg
In our first plenary session of the afternoon, we were delighted to welcome Professor Steven Salzberg, Professor and Director of the Centre for Computational Biology at Johns Hopkins University. Professor Steven Salzberg shared his team’s recent work developing hybrid assembly methods that utilise a combination of short and long reads to deliver high-quality, highly contiguous genome assemblies.
In essence, the assembly method works by taking short reads and condensing them down to deliver ‘super-reads’, which are then mixed with long nanopore reads to create ‘mega reads’ – with the long reads acting as a scaffold during the alignment.
The team at Johns Hopkins University have applied this new assembly strategy to study the English walnut tree (600Mb). Based on short-read sequencing alone, they obtained a contig N50 of 46,148 bp; however, adding in the long reads increased this to 1,324,206 bp. Using the nanopore platform, the team generated 7 million reads, representing 35x genome coverage.
Based on the success of this project, further organisms have now been analysed using the hybrid approach, including the pathogenic fungi Lomentospora prolificans, Trichosporon mucoides and Trichosporon dermatis. Interestingly, the T. mucoides, genome was approximately twice as large as anticipated, which Professor Salzberg hypothesizes may be caused by heterozygosity in this diploid fungus.
Professor Salzberg also touched on his team’s role assembling the extremely challenging genomes of two of the largest trees on Earth: the giant sequoia, Sequoiadendron giganteum(11 Gb) and the Coast Redwood, Sequoia sempervirens (33 Gb). Professor Salzberg revealed that the sequoia sequencing has now finished and assembly is in progress.
The hybrid assembly algorithms described in this presentation have been integrated into the MaSuRCA system, which, as with the other analysis tools developed in Professor Salzberg’s lab, such as Bowtie, Tophat, and Cufflinks, is freely available to download.
Steven Salzberg was interviewed after his plenary:
Clinical panel
Justin O’Grady
Justin O’Grady, Associate Professor of Medical Microbiology at the University of East Anglia, started the session with a question: ‘In a world of rising antimicrobial resistance (AMR) is culture-based diagnosis fit for purpose?’ He believes it is not, as it is ‘too slow’. Justin is pioneering this use of nanopore sequencing to deliver improved pathogen identification.
Justin described his latest research utilising the MinION to develop a rapid metagenomic test for hospital-acquired pneumonia (HAP). HAP, where respiratory infection develops over 48-hours after hospital admission, affects 1.5% (200k) of UK hospital inpatients per year, with a 25-50% mortality rate associated with infection. Justin outlined how current culture-based diagnostics not only have poor sensitivity and specificity, but are also too slow to impact patient management. To combat this, Justin and his team have developed a metagenomic workflow which includes human DNA depletion, pathogen DNA extraction, library preparation, MinION sequencing and EPI2ME analysis. Validating this workflow on over 50 respiratory samples, the team were able to go from sample to resistance gene identification in approximately 6 hours — a timeframe allowing improved patient management. This compares extremely favourably with the 48 hours required for traditional culture-based techniques. Overall, the metagenomic approach was found to be 92% concordant with standard microbiology testing.
Further analysis of resistance data is in progress; however, Justin revealed that it was possible to detect the mecA gene in all MRSA positive samples.
Summarising, Dr O’Grady stated that: ‘Real-time metagenomics has the potential to replace culture for the diagnosis of pneumonia and provides the rapid turnaround necessary for precision management of HAP patients’.
Alban Ramette
Our second panel member, Dr Alban Ramette leads the Bioinformatics/Biostatistics group at the University of Bern, with a focus on high-throughput sequencing solutions for clinical applications. Alban started his presentation by pointing out that PCR, the most commonly used technique for routine viral identification in a clinical setting, misses many genetic aberrations (e.g. point mutations and recombination events), unless followed up by an additional approach such as Sanger sequencing. As such, PCR assays alone may lead to false positive results. Alban elaborated on his group’s latest research that aims to address these challenges through the use of nanopore sequencing. Alban set out to discover if nanopore sequencing would allow them to identify enteroviruses (EV) strains in culture or directly from native samples.
The team evaluated both direct RNA sequencing and indirect RNA sequencing (cDNA).
Using cDNA sequencing, the team were able to obtain >95% coverage of EV genomes with 99% consensus accuracy. Alban stated that the level of sensitivity delivered is equivalent to PCR. The data also revealed that, when sequencing cDNA, there is no further appreciable benefit in accuracy after 5-10 minutes of the sequencing run, especially when the consensus sequences based on the first reads are further improved via nanopolish.
Alban also reported his results of using direct RNA sequencing, which excitingly provided nearly the full RNA genome in a single sequence, allowing easier analysis of genomic rearrangements and quantitation in native samples.
He summarised by stating that cDNA nanopore sequencing works really well with high accuracy, while direct RNA-seq offered the fastest turnaround from sample to result and has tremendous potential for pathogen identification in a clinical setting.
Claire Jenkins
Our final panel member, Dr Claire Jenkins, Deputy Head of the Gastrointestinal Bacteria Reference Unit at Public Health England (PHE), shared her work investigating the potential for integrating the MinION into routine service for typing Shiga Toxin producing E. Coli (STEC).
Claire presented the results of a pilot study, which demonstrated how nanopore whole genome sequencing data could be incorporated into the existing PHE bioinformatics pipelines and integrated with short-read data.
Claire revealed that the long reads delivered by the MinION allowed more accurate characterisation of prophage than provided by short read data, which generated many false positives. More accurate prophage characterisation may provide important clues to the source and transmission of this clinically significant zoonotic, foodborne pathogen.
Summarising, Claire stated that they plan to continue running short and long-read sequencing in parallel for routine surveillance and outbreak investigations. They are also aiming to develop nanopore sequencing protocols for analysis of Vibrio and Yersinia species, both for routine surveillance and rapid outbreak response.
Plenary: the Nanopore Human RNA Consortium
Miten Jain and Winston Timp
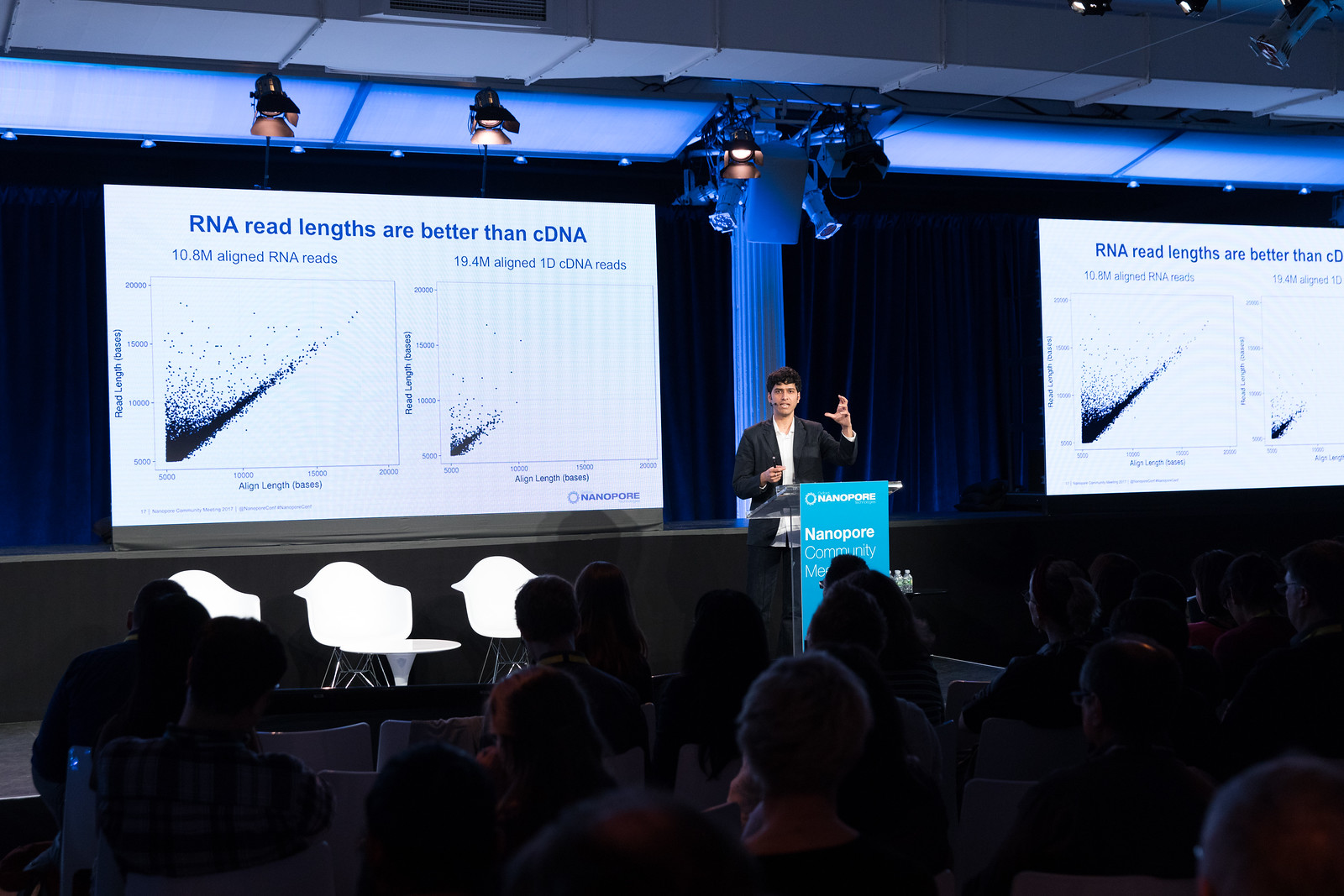
First up, Winston Timp from Johns Hopkins University set the scene for this multicentre project involving six teams located across the UK, Canada and Europe. The aim of the study is to sequence the transcriptome of NA12878 using direct RNA and cDNA approaches on the nanopore platform.
In their hands direct RNA had marginally higher accuracy of 87% in comparison to cDNA which had an accuracy of 86%; however, cDNA sequencing higher throughput. The team also found that 750 ng input RNA provided higher numbers of reads (1 million), than the 500 ng typically recommended.
Miten Jain from UCSC then came to the stage to explain why he believes nanopore sequencing of RNA is so important, which included:
• Long reads provide exon connectivity
• Due to the read-length, every read gives an isoform prediction
• RNA reads can provide accurate measurement of gene fusions
• It allows an estimation of poly-A length
• Detection of RNA modifications
Closing the talk, Miten announced that the preliminary data from this study is now available on GitHub, encouraging the research community to help maximise the impact and drive forward this initiative.
Miten and Winston were interviewed after their talk:







