Direct-from-colony microbial sequencing: rapid Salmonella serotyping know-how and info sheet

Requirements
Direct-from-colony microbial sequencing: rapid Salmonella serotyping know-how and info sheet
FOR RESEARCH USE ONLY.
Contents
Introduction
Sample input considerations
Library preparation – considerations for optimal performance
Sequencing set up and run parameters
*Salmonella* serotyping
- 4. *Salmonella* 24-plex run direct from cell colony
- 5. Testing closely related *Salmonella* serotypes
Different run strategies
Flow cell wash and reuse
Disclaimers
Introduction
Salmonella spp. are the most common aetiological agents of food borne illness with over a million people a year affected in the U.S. alone, ~ 26,500 of which are hospitalised, resulting in ~420 deaths 1-3. Outbreaks of salmonellosis can result from many kinds of foods: testing and tracing outbreaks and origins is of great interest to government, healthcare, and food industry alike. Whole genome sequencing (WGS) of isolated Salmonella organisms is becoming the gold standard for molecular characterisation and a number of approaches may be taken to achieve this 4-5.
We have developed a protocol that describes the methodology and analysis steps in detail:
This method uses our Rapid PCR Barcoding Kit (SQK-RPB114.24) to provide a simple end-to-end solution to generate whole genome sequencing data directly from a single Salmonella colony, eliminating the need for subculture into liquid broth, DNA extraction or complex library preparation methods, thereby offering faster tun-around times (Figure 1). This is combined with our custom wf-bacterial-genomes analysis pipeline using EPI2ME to provide an organism species ID, serotype, multi locus sequence type (MLST) and antibiotic resistance (AMR) profile information 6. This method is designed to provide non-circular genomes for research and routine lab operations, including food safety testing purposes.
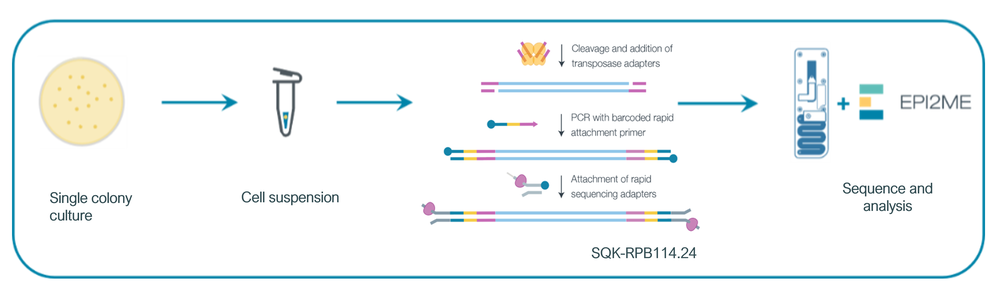 Figure 1: End-to-end workflow for rapid WGS of Salmonella spp.
Figure 1: End-to-end workflow for rapid WGS of Salmonella spp.
Introduction references
- https://www.cdc.gov/salmonella/index.html
- https://www.fda.gov/animal-veterinary/animal-health-literacy/get-facts-about-salmonella
- https://www.who.int/news-room/fact-sheets/detail/food-safety
- Annu. Rev. Food Sci. Technol. 2016, 7:353–74
- Front. Microbiol. 2023, 13:1073057. doi: 10.3389/fmicb.2022.1073057
- https://labs.epi2me.io/workflows/wf-bacterial-genomes
Sample input considerations
The input requirements for this end-to-end workflow are single colonies of Salmonella and allow multiplexing of up to 24 samples on a single flow cell.
Colonies of Salmonella enterica were grown overnight (~16-18 h) on agar plates. Single colonies were picked and resuspended for use in the sample preparation. For optimal downstream processing and sequencing, a PCR yield of at least 15-20 ng/µL is required. An estimation of the impact of colony size on PCR yields was carried out (Figure 2). We recommend picking colonies with a diameter of ~2-3 mm: colonies larger or smaller than this typically did not produce the required yield of DNA.
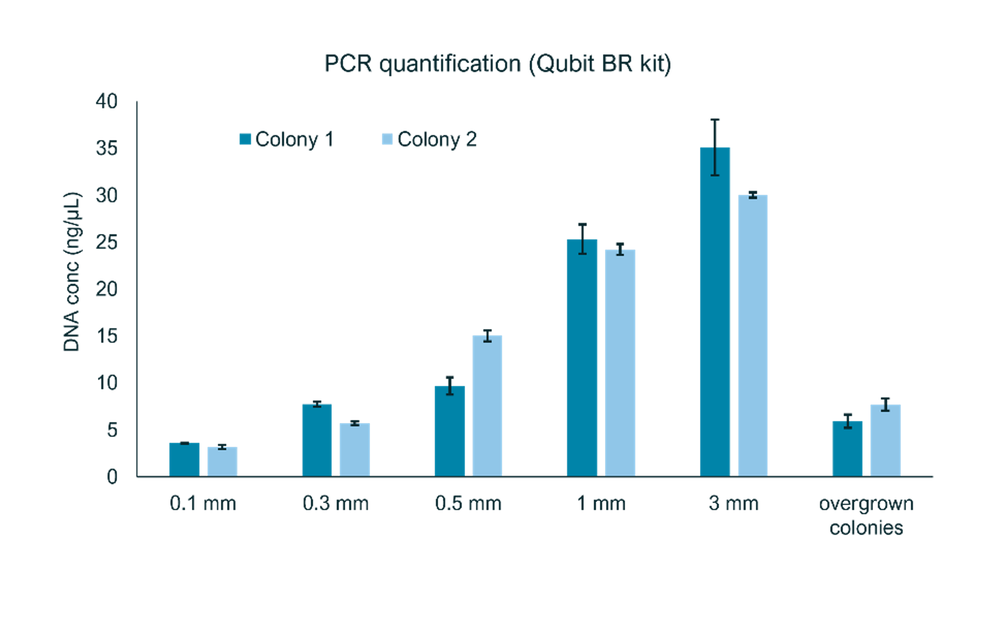
Figure 2: Screening the best colony size for the end-to-end workflow.
Colonies with size 2-3 mm gave the best PCR amplification when performed with 20 cycles using 6 minutes of extension (see following section for more information). Colonies <1 mm in size did not amplify well and did not produce sufficient DNA for sequencing. Similarly, overgrown colonies did not amplify well.
An important consideration when picking colonies is to ensure that no growth medium (agar) is scraped up. Growth medium or agar can interfere with the PCR reaction and result in poor PCR amplification yields.
Escherichia coli (E.coli) was studied as a surrogate for Salmonella. Both E.coli and Salmonella being gram negative micro-organisms and, having observed equivalent performance of E.coli colonies with Salmonella colonies in PCR reactions, all further protocol optimisations were done using E.coli colonies.
Library preparation – considerations for optimal performance
PCR cycle number
PCR reactions were set up using the same tagmented DNA pool from a single E.coli colony and tested across three different barcodes (Figure 3). Water was used as negative control. PCR setup is described in the figure below.
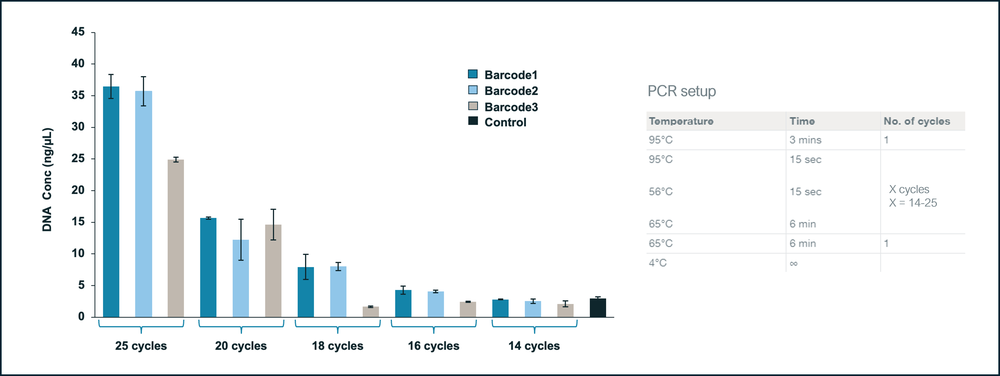
Figure 3. Optimising PCR cycle number.
Thermocycling for 25 cycles gave the best PCR amplification yields when using the same tagmented pool of DNA from a single colony across various barcodes.
Based of the above results, we recommend using 25 cycles for PCR amplification to achieve optimal performance.
Polymerase extension time
The extension time of LongAmp Taq polymerase was optimised to determine the optimal time required for the best performance in terms of coverage and barcode balance. PCR reaction was set up using the same tagmented DNA pool from a single E.coli colony and tested across three different barcodes (Figure 4). PCR was set up with varying extension times from 2-6 minutes. 6 minute extension leads to longer sequence lengths being obtained (Figure 4a) although there was not much of an impact on coverage between 4 and 6 minute extension times (Figure 4b).
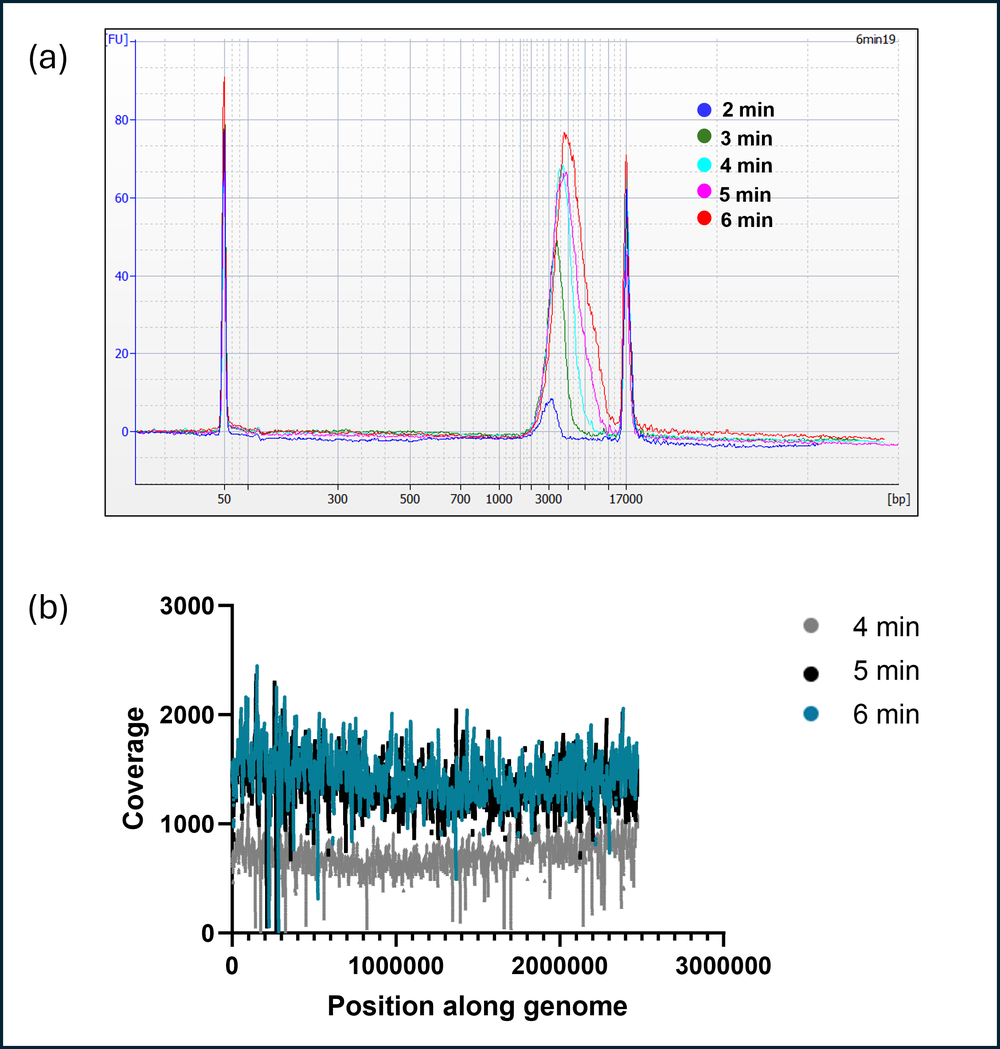
Figure 4. Optimising PCR extension time.
(a) Sequence lengths of amplicons from the PCR reaction using different extension times (b) Depth of coverage across the genome. The results indicated that 6 min extension gave longer amplicons, and better barcode distribution.
Sequencing set up and run parameters
We recommend that sequencing is carried out until the lowest represented barcode has at least 50k reads. Following internal testing, we recommend various sequencing times depending on the number of barcodes used:
Table 1. Recommended sequencing times using SQK-RPB114.24 kit for WGS of Salmonella spp.

*Salmonella* serotyping
*Salmonella* 24-plex run direct from cell colony
24 individual colonies from a plate of known Salmonella serotype were processed following the protocol. Analysis of the data using parameters described in the protocol analysis section showed correct serotype prediction within 4 hours of sequencing (Figure 5). At 4 hours of sequencing, the average number of reads was approximately 15,000 per sample resulting in ~15X coverage.
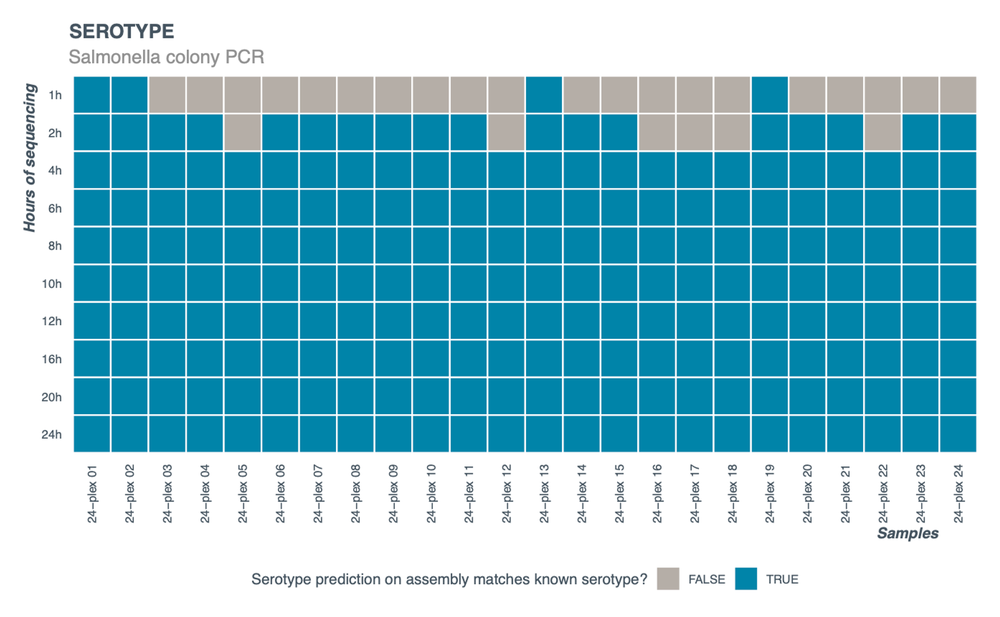
Figure 5. Serotype prediction using SeqSero2 (see Data analysis section of the protocol for details) of a 24-plex run from 24 individual Salmonella colonies of known serotype.
Data was extracted at different timepoints into the sequencing run.
Testing closely related *Salmonella* serotypes
We prepared and sequenced genomic DNA from ten different Salmonella serotypes ordered from Cornell University (barcodes 3, 5, 7, 9, 11, 14, 17, 19, 21 and 23). These samples are known to be closely related serotypes. The samples were run alongside other Salmonella samples as a 24-plex run.
A comparison of the predicted serotype with the expected serotype showed that all samples had correct and final serotype prediction by 6 hours of sequencing (Figure 6). At 6 hours of sequencing, the average number of reads was approximately 35,000 per sample resulting in ~17X coverage. In the first few hours of sequencing, for two samples (barcodes 07 and 17), the serotype prediction was unstable (as indicated by the blue turning grey) – however, the predicted serotypes and antigenic profiles remained close to the final serotype call across all time points.
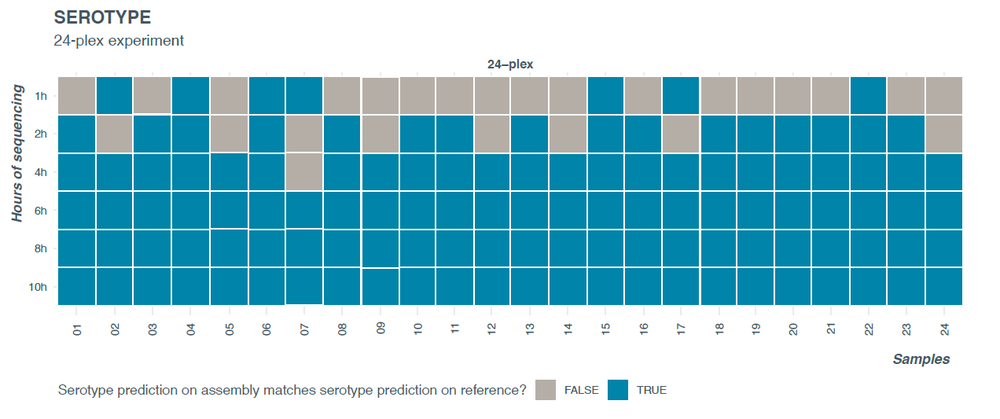
Figure 6. Serotyping closely related Salmonella serotypes using SeqSero2.
Barcode 07 types as I -:z:1,6 (antigenic profile -:z:1,6) at 2-4h and as Poona (antigenic profile 13:z:1,6) at other timepoints. Barcode 17 types as - 1,3,19:d:e,n,z15 (antigenic profile 1,3,19:d:e,n,z15) at 2 h and as Liverpool (antigenic profile 1,3,19:d:e,n,z15) at other timepoints.
Different run strategies
Multiplexing
Up to 24 barcodes are available to multiplex with the SQK-RPB114.24 kit and the user can choose a different level of multiplexing depending on the number of samples and time available to sequence. As the number of samples multiplexed on a flow cell decreases, each barcode accumulates more reads within the same run time, reducing the sequencing time needed to correctly serotype. Figure 7 shows serotyping results at different sequencing timepoints for a 6-plex and 12-plex runs prepared from different input Salmonella serotypes (genomic DNA). All samples had correct serotype prediction by 4 hours of sequencing. At 4 hours of sequencing, the average number of reads for the 6-plex run was approximately 100,000 reads per sample resulting in ~45X coverage. The 12-plex run had average of 50,000 reads and ~25X coverage at 4 hours.
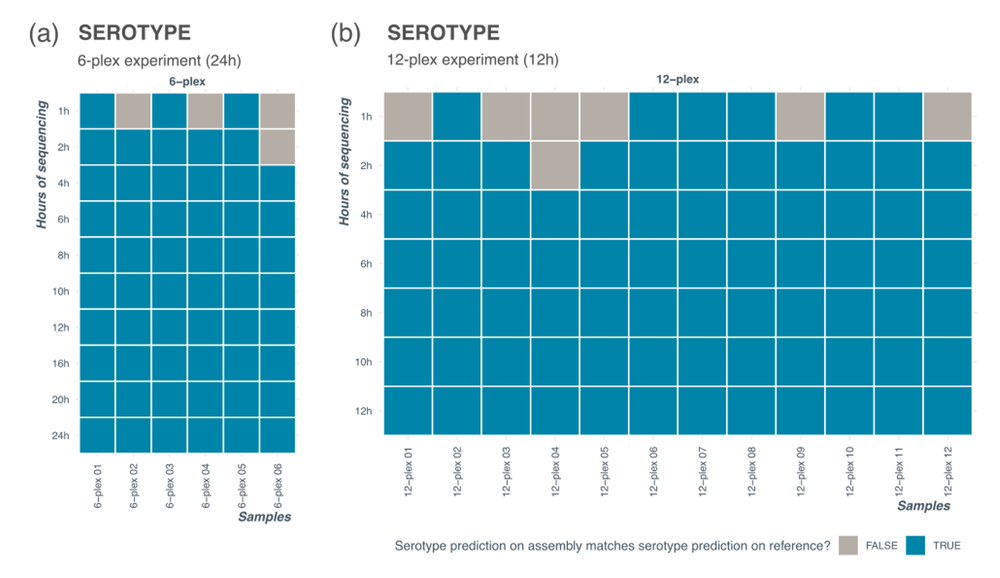
Figure 7. Serotyping results for levels of multiplexing.
Serotyping results for a 6-plex (a) and a 12-plex (b) experiment where data was extracted at different timepoints into the sequencing run. Correct serotype was established by running serotyping analysis tool (SeqSero2 – see Data analysis section of the protocol for details) on reference assembly available from commercial sample provider.
Flow cell wash and reuse
If a subset of barcodes (example 6 or 12 barcodes) is used, it is possible to achieve sequencing in a shorter run time. Hence, the flow cell could be reused with a new set of barcodes thus maximizing flow cell utility.
In order to assess the effect of washing the flow cell and reloading the same flow cell with a separate set of barcodes, we prepared two sets of samples:
- Library 1 with barcodes 1–12
- Library 2 with barcodes 13–24
Sequencing with Library 1 (barcodes 1–12) was carried out for 8 hours, following which the flow cell was stopped and washed using the Flow Cell Wash Kit (EXP-WSH004). Following the wash, the flow cell was reloaded with Library 2 (barcodes 13-24) and sequenced for 72 hours (Figure 8). Analysis showed less than 1% carryover of Library 1 barcodes into Library 2 sequencing. Further, there was no visible impact of sequence length distribution post-wash, which indicated that the wash protocol did not have any detrimental effects on the second library being loaded onto flow cells (Figure 9).
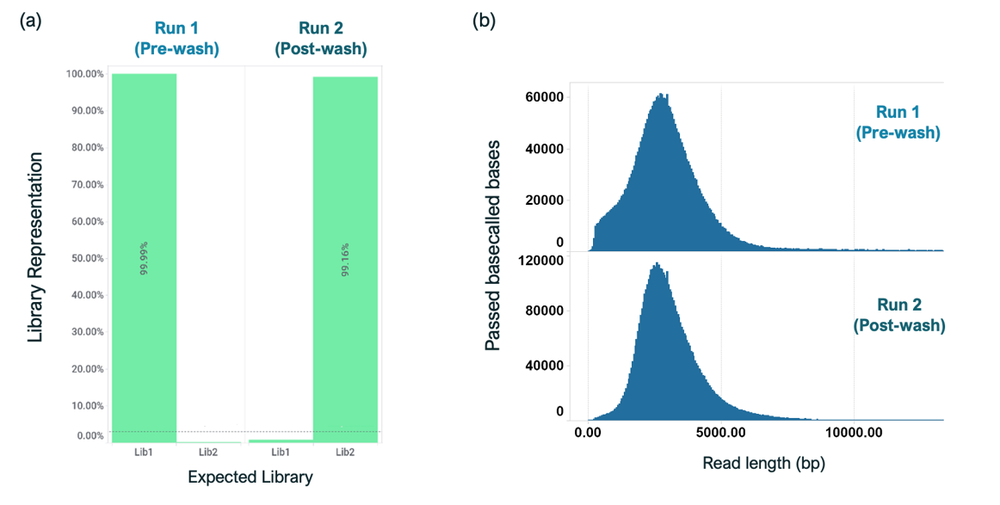
Figure 8. Assessing the impact of wash and reload in the sequencing runs.
(a) Analysis of library carryover post wash indicated that 0.84% of Library 1 was present in Library 2. (b) There was no significant impact on read lengths of Library 2 loaded post wash.
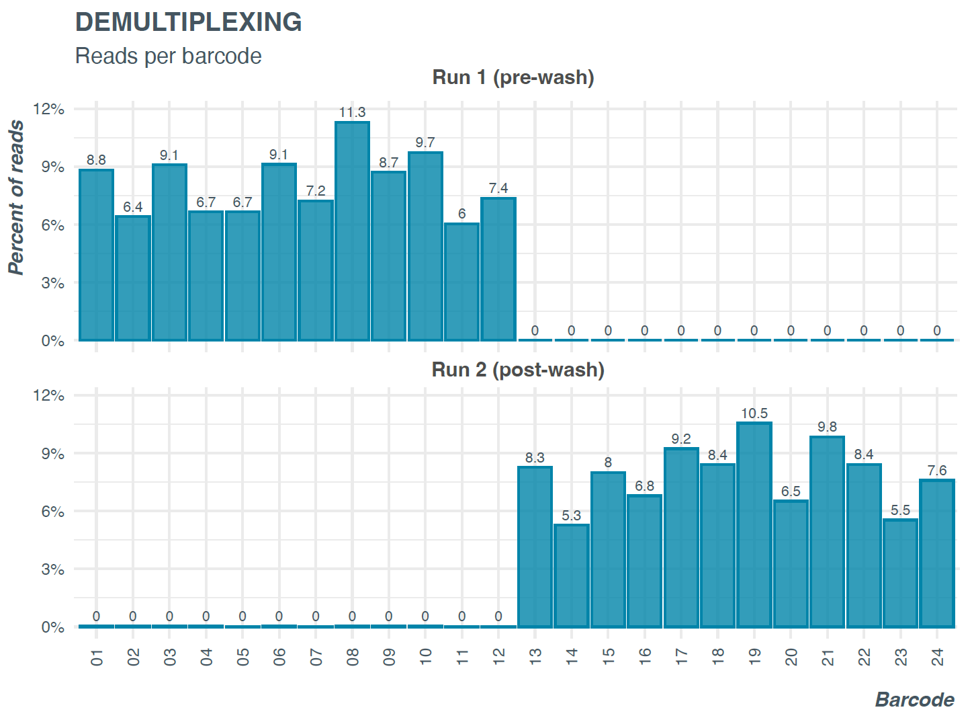
Figure 9. Barcode distribution before and after wash and reload on same flow cell.
Run 1 shows barcode distribution of Library 1 (barcode 1-12, before wash) and Run 2 shows barcode distribution of library 2 (barcode 13-24) loaded on to the same flow cells after washing using the wash kit EXP-WSH004. Very few reads (<1%) from barcodes 1-12 are detected after wash and reload.
Serotyping results for the runs before and after wash and reload can be seen in Figure 10. Correct serotyping is achieved by 8 hours of sequencing in both cases. At 8 hours of sequencing, the average number of reads was approximately 80,000 (Library 1, pre-wash) and 60,000 (Library 2, post-wash) per sample resulting in ~30X (pre-wash) and ~20X (post-wash) coverage respectively.
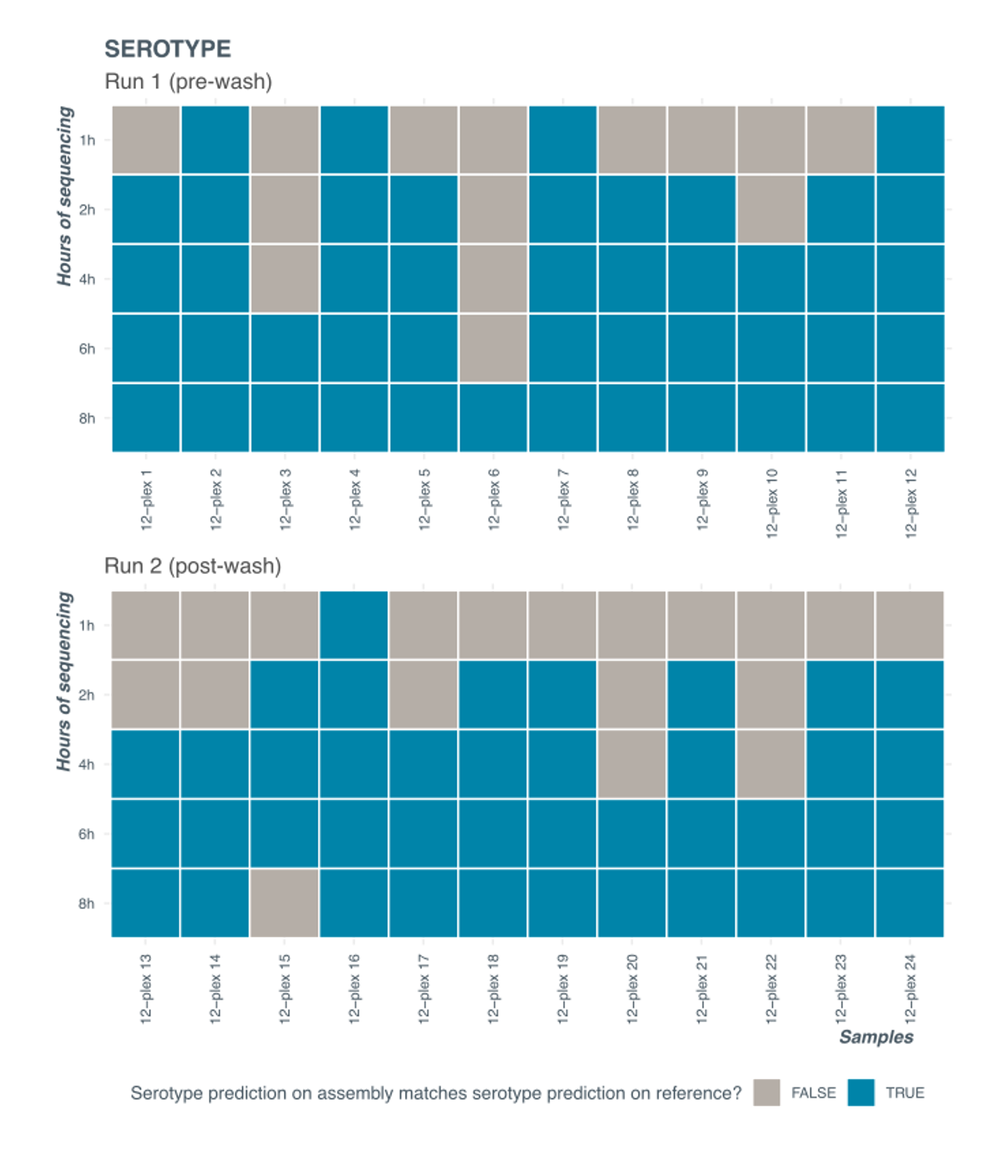
Figure 10. Serotyping results for a wash and reload experiments.
Data was extracted at different timepoints into the sequencing run. Correct serotype was established by running serotyping analysis tool (SeqSero2 – see Data analysis section of the protocol for details) on reference assembly available from commercial sample provider. Barcode 15 types as VI 45:z4,z23:e,n,x (antigenic profile 45:z4,z23:e,n,x) at 8h and as VI 45:a:e,n,x (antigenic profile 45:a:e,n,x) at 6 hours.
Disclaimers
Following NARMS Manual of Laboratory Methods (fda.gov), SiSTR and NCBI or outsourcing to a reference laboratory can help assign a final consensus serotype for results that are inconclusive using SeqSero2. Per Foods Program Compendium of Analytical Laboratory Methods | FDA, SeqSero2 is the recommended pipeline by FDA and USDA FSIS for whole genome sequencing of Salmonella isolates. If the sample serotype is not called after the recommended sequencing time, and the sample is known to be Salmonella, users can follow instructions in the protocol for reanalysis using kmer mode in SeqSero2.
Change log
| Version | Change |
|---|---|
| v1, Sept 2024 | Initial publication |






