Resolving translocations in the context of PGD using long nanopore sequencing reads
Structural variants (SVs) are known to be important in genetic diseases, by damaging or changing the functions of important genes. Chromosomal translocations, a class of SVs, can damage normal gene expression and function. However, typical methods of identifying translocations, such as karyotyping, fluorescence in situ hybridization (FISH), and Southern blot, are not sensitive enough to precisely identify the translocation breakpoints, and so the impact of the translocation on gene structure and function is often unknown.
The advantages of long-read sequencing for identifying translocations
Short-read sequencing can help detect translocations and identify breakpoints more precisely, but when breakpoints are located in repeat-rich regions it is difficult to accurately identify their location. Long-read sequencing can greatly improve SV detection, regardless of whether or not an SV is in a repetitive region. Long reads are also helpful for resolving haplotypes between translocations and nearby SNPs, which could be particularly important in preimplantation genetic diagnosis (PGD): balanced translocations occur in ~0.2% of the human population and in 2.2% in patients with a history of recurrent miscarriages or repeated in vitro fertilization failure.
Recognising the importance of identifying translocation breakpoints in the context of PGD, we used nanopore sequencing to detect translocations and precisely define their breakpoints, in individuals with and without long-standing infertility problems. Translocations had initially been detected by conventional karyotyping. We also obtained haplotype information.
Our laboratory and analysis workflows
We investigated translocations in seven individuals, three females and four males; three of which had long-standing infertility. Among them, six balanced translocations and one inversion had been previously identified by karyotyping. We extracted their genomic DNA and prepared it for nanopore sequencing using the Ligation Sequencing Kit; the libraries were then sequenced on the GridION.
To identify SVs, we used an analysis pipeline that combined NGMLR-sniffles and LAST-NanoSV, and for haplotyping, we used MarginPhase. We verified the translocation breakpoints with PCR, and Sanger sequencing of the amplified products.
Detecting and characterizing the translocation breakpoints
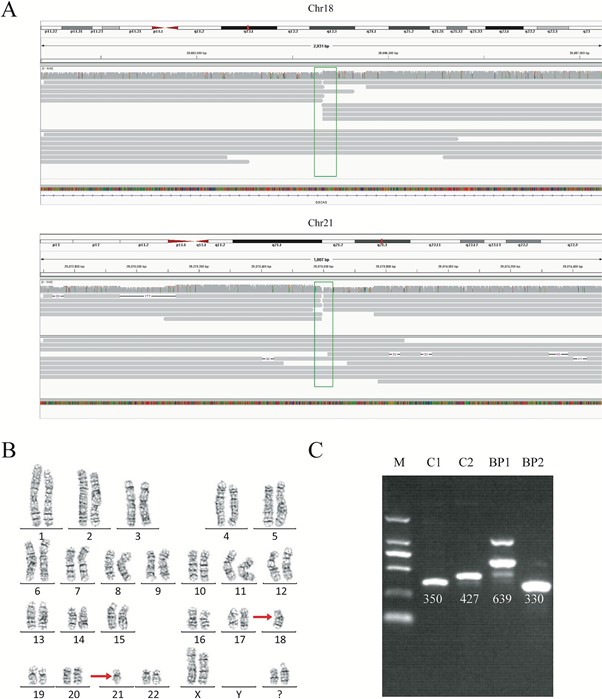
For each genome, we obtained 32-44 Gb of sequence data, with a mean read length of 12.3-16.3 kb and a depth of 9.9-13.5x. With our analysis pipeline we successfully discovered 14 breakpoints in the seven individuals, and the breakpoint locations were consistent with the karyotyping results (Figure 1). Around 10 reads covered each breakpoint.
By viewing the breakpoints in the UCSC Genome Browser, we found breakpoints inside introns of genes CSMD3, AK129567, AK302545, RNF139, and CCDC102B, in four individuals. Therefore, the structures of these genes were significantly disrupted, as a portion of each gene had moved to another chromosome. Interestingly however, there was no obvious impact on the phenotype of these four carriers, except for primary infertility. We also found microdeletions and insertions in conjunction with the translocations in two carriers, although the mechanisms behind these remain unknown. We also found that in three cases, the breakpoints occurred in repetitive Alu or LINE elements.
Interestingly, we found that in one individual with a karyotype of 46, XX, t(3;9) (p13;p13), the breakpoint on chromosome 3 was very close to the acrocentric centromere. Parts of the long reads that supported the breakpoint in chromosome 9 could be mapped, but due to a gap in the reference genome (hg19) at this locus, the position of the breakpoint was imprecise. However, the long reads showed strong evidence that the breakpoint was in the centromere, demonstrating how long reads have the ability to detect breakpoints in such low complexity regions of the genome.
Inversions, like translocations, can also be difficult to detect with short-read sequencing. With long nanopore reads, we successfully detected an inversion in one carrier, and this was verified with PCR and Sanger sequencing.
Breakpoint validation
We wanted to validate the exact translocation breakpoints detected with nanopore sequencing, and so performed PCR and Sanger sequencing of the breakpoints we had identified. Validation was successful in four samples of the seven. In the other three, it was challenging to obtain a PCR product, despite multiple attempts, because the breakpoints were in highly repetitive regions. This showed us the power of long-read sequencing to precisely detect translocation breakpoints in low complexity regions, compared to other methods.
Haplotyping the structural variants
As we know that haplotype identification is important in PGD, we also investigated this in our cases. We successfully detected informative SNPs near the translocation breakpoint regions, and this enabled haplotyping of the chromosomal regions involved (Figure 2). This was possible from only 10x depth of coverage.

Conclusions
In this research, we successfully identified and sequenced every breakpoint in our seven carriers, using nanopore sequencing. All breakpoints were consistent with their corresponding karyotype results. We also found that in four cases the breakpoints were located in repetitive regions, showing how long sequencing reads are able to analyse even highly repetitive and complex regions.
We suggest that low-coverage, whole-genome sequencing using nanopore technology is a powerful tool for precisely locating translocation breakpoints. In future, long-read nanopore sequencing may play an important role in analysing chromosomal translocations in the context of PGD and assisting reproduction and preimplantation decisions.


This work was undertaken by Liang Hu’s team (left), at the Reproductive and Genetic Hospital of Citic-Xiangya, and their collaborators at GrandOmics (right).
L. Hu et al. Location of balanced chromosome-translocation breakpoints by long-read sequencing on the Oxford Nanopore platform. Frontiers in Genetics. DOI: https://doi.org/10.3389/fgene.2019.01313 (2020).






