Blog: Metagenomic sequencing: which assembly method is best?
In this blog, Adriel Latorre-Perez shares his work on comparing methods for assembling metagenomes from nanopore sequencing data, and he provides recommendations on the best tools to use for the job.
 Adriel Latorre-PerezI am a bioinformatician with a special interest in third generation sequencing and metagenomics. I am also a PhD student at Darwin Bioprospecting Excellence, where I’ve been working with nanopore data since 2017. The main objectives of my research are (1) to evaluate the suitability of ONT sequencers for different metagenomic approaches, and (2) to apply this technology for studying environments of industrial or biotechnological interest. In the last few years, we have successfully applied MinION for characterizing microbial communities associated with biogas production processes, via 16S rRNA sequencing (Archaea and Bacteria). At this moment, we are also evaluating and optimizing different methodologies for metagenomic analysis. Mainly, we are focused on benchmarking 16S rRNA pipelines and metagenomic assembly tools. In fact, I am developing a GitHub repository, which includes the results and the supplementary information of our recent work “Assembly methods for nanopore-based metagenomic sequencing: a comparative study”. The repository is intended to be a dynamic source of benchmarking results for different assembly tools and datasets, with the aim of helping other researchers to select the most appropriate tool for their specific application.
Adriel Latorre-PerezI am a bioinformatician with a special interest in third generation sequencing and metagenomics. I am also a PhD student at Darwin Bioprospecting Excellence, where I’ve been working with nanopore data since 2017. The main objectives of my research are (1) to evaluate the suitability of ONT sequencers for different metagenomic approaches, and (2) to apply this technology for studying environments of industrial or biotechnological interest. In the last few years, we have successfully applied MinION for characterizing microbial communities associated with biogas production processes, via 16S rRNA sequencing (Archaea and Bacteria). At this moment, we are also evaluating and optimizing different methodologies for metagenomic analysis. Mainly, we are focused on benchmarking 16S rRNA pipelines and metagenomic assembly tools. In fact, I am developing a GitHub repository, which includes the results and the supplementary information of our recent work “Assembly methods for nanopore-based metagenomic sequencing: a comparative study”. The repository is intended to be a dynamic source of benchmarking results for different assembly tools and datasets, with the aim of helping other researchers to select the most appropriate tool for their specific application. In recent years, shotgun metagenomic sequencing has become a powerful tool for recovering individual genomes directly from complex microbiomes, leading to the identification and description of new, relevant – and mainly unculturable – taxa, with meaningful implications. When performing de novo assembly, using short reads often results in highly fragmented genomes, even if sequencing is carried out from pure cultures. This is a consequence of the inability to correctly assemble genomic regions containing repetitive elements that are longer than read length (Goldstein et al., 2019). The fragmentation problem is magnified when handling metagenomic sequences due to the existence of intergenomic repeats. Moreover, microbial communities often contain related species or sub-species in different – and unknown – abundances, resulting in extensive intergenomic overlaps that make the global assembly especially difficult (Ayling et al., 2019).
Oxford Nanopore sequencing platforms have recently emerged as a solution to resolve ambiguous repetitive regions and to improve genome contiguity. Despite their relatively high error per sequence, their ability to produce long reads has allowed them to generate assemblies with a high degree of completeness (Goldstein et al., 2019, Wick et al., 2017). These promising results have favoured the development of a range of assemblers intended to deal with nanopore data, in order to obtain better and more contiguous assemblies.
Addressing the uncertainty of selecting a genome assembly tool
The rapid appearance of new tools may lead Oxford Nanopore users to an uncertainty about which analysis software to choose for their specific applications, and thus benchmarking studies are needed to systematically evaluate the comparative performance of the different available tools. We found that the evaluation studies made for nanopore-based shotgun sequencing data had focused on reconstructing single bacterial genomes from isolates, but not metagenomes (Goldstein et al., 2019; Tyler et al., 2018; Sović et al., 2016).
For that reason, we decided to use the data from Nichols et al. (2019) for the benchmarking of different available assembly tools. Data consisted of the ultra-deep nanopore sequencing of two different mock communities with GridION and PromethION platforms. Both communities included the same species (eight bacteria; two yeasts), but differed in the expected proportion for each microorganism. As we wanted to study the suitability of MinION – the most used Oxford Nanopore sequencing platform – to reconstruct individual microbial genomes from metagenomes, we subsampled the GridION and PromethION datasets to have a final output of approximately 3 and 6 Gb, which we determined to be the typical MinION output at the time of the analysis.
Comparing assembly tools: completeness, accuracy, and time
After installing and running the different assemblers, MetaQUAST was employed for evaluating the degree of completeness of the individual species draft genomes. Overall, Flye and Canu performed the best (Figure 1). These tools were able to reconstruct all of the bacterial genomes with a high percentage of completeness. Flye slightly outperformed Canu in terms of individual genome recovery, since it was able to reconstruct a significantly higher percentage of some genomes (Cryptococcus neoformans, Saccharomyces cerevisiae and Staphylococcus aureus).
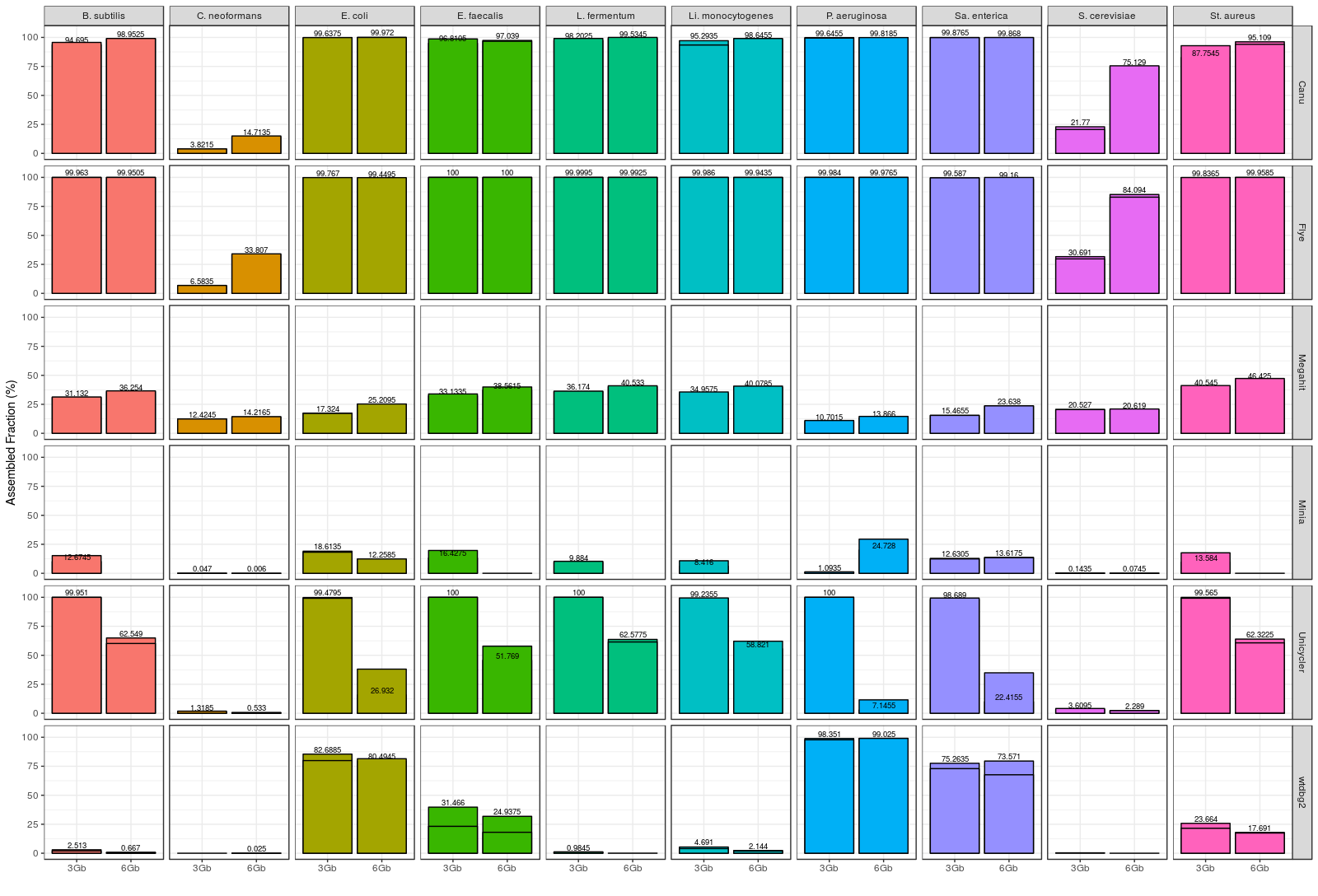
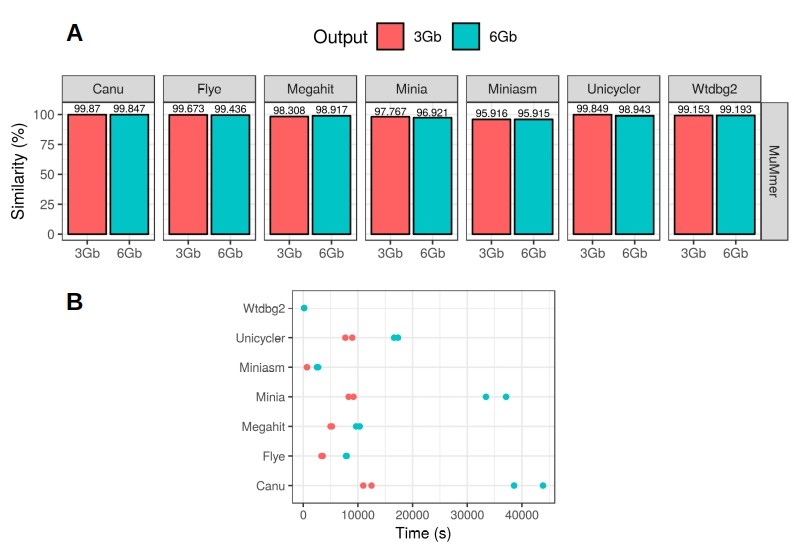
We also evaluated the accuracy of the results by aligning the different draft metagenomes to a high quality reference with MuMmer4. Canu and Flye were found to be the most accurate assembly tools, but this time Canu slightly outperformed Flye (Figure 2A). In fact, Canu was able to achieve up to 99.87% consensus accuracy, demonstrating that nanopore sequencing errors can be easily corrected after the assembly of the reads. Finally, we registered the time used by each tool for generating the final assembly (Figure 2B). Wtdbg2 was the fastest tool, followed by miniasm, and then Flye. Canu was, in fact, the slowest assembler, taking more than 10 hours to complete the 6 Gbp assemblies. Full results and discussion can be found in the Latorre-Pérez et al. (2019) BioRxiv article.
Assembly tool recommendations
In light of our results, we would recommend using Flye for general metagenomics applications (Figure 3). This tool led to the highest metagenome recovery ratio and performed in a fast and robust way among all the tested datasets. Canu would be more suitable when lower error rates are required, as in the case of biosynthetic gene cluster prediction.
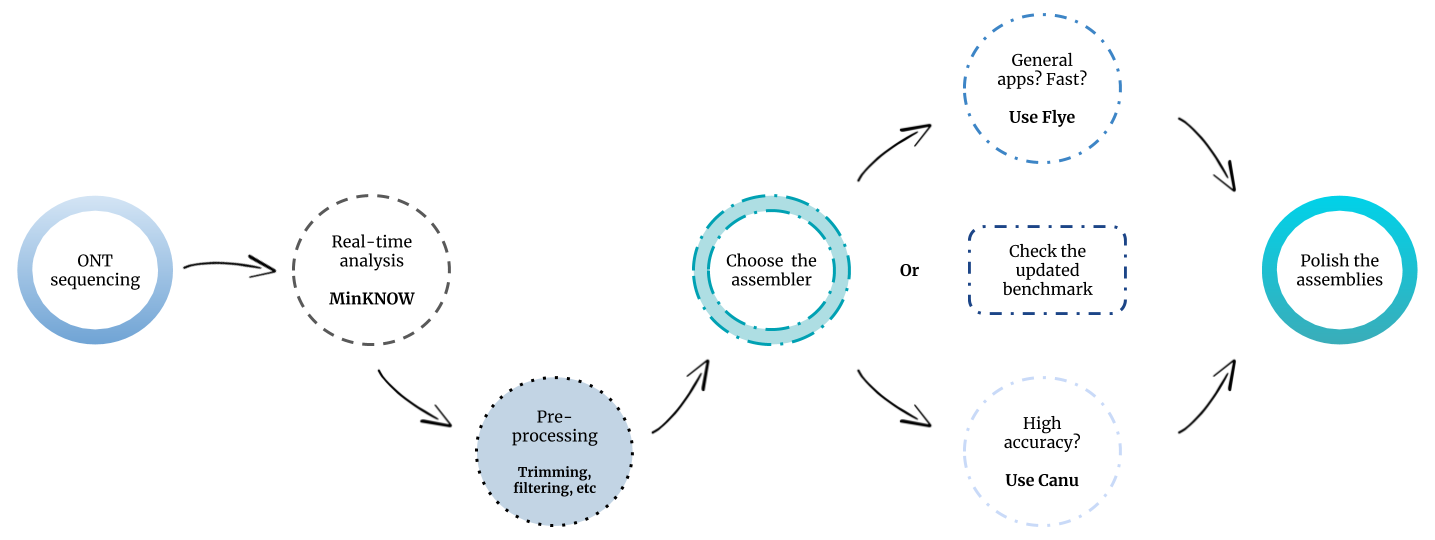
It has to be highlighted that the development of nanopore sequencing data analysis tools is a highly dynamic process, in which new assemblers or new versions of existing assemblers are constantly published. Shasta, Raven, Pomoxis and MetaFlye (Flye v.2.7) are examples of new software/versions which were not available at the time that we carried out the experiments. Moreover, the output of the MinION is constantly increasing, commonly achieving up to 15-20 Gb per flow cell at this time. For that reason, we decided to create a GitHub repository, with the intention of periodically updating our evaluation study with new assembly tools and data.
References
- Goldstein, S., Beka, L., Graf, J. and Klassen, J. (2019). Evaluation of strategies for the assembly of diverse bacterial genomes using MinION long-read sequencing. BMC genomics, 20(1).
- Ayling, M., Clark, M. and Leggett, R. (2019). New approaches for metagenome assembly with short reads. Briefings in Bioinformatics.
- Wick, R., Judd, L., Gorrie, C. and Holt, K. (2017). Completing bacterial genome assemblies with multiplex MinION sequencing. Microbial Genomics, 3(10).
- Tyler, A., Mataseje, L., Urfano, C., Schmidt, L., Antonation, K., Mulvey, M. and Corbett, C. (2018). Evaluation of Oxford Nanopore’s MinION Sequencing Device for Microbial Whole Genome Sequencing Applications. Scientific Reports, 8(1).
- Sović, I., Križanović, K., Skala, K. and Šikić, M. (2016). Evaluation of hybrid and non-hybrid methods for de novo assembly of nanopore reads. Bioinformatics, 32(17), pp.2582–2589.
- Nicholls, S., Quick, J., Tang, S. and Loman, N. (2019). Ultra-deep, long-read nanopore sequencing of mock microbial community standards. GigaScience, 8(5).
- Latorre-Pérez, A., Villalba-Bermell, P., Pascual, J., Porcar, M. and Vilanova, C. (2019). Assembly methods for nanopore-based metagenomic sequencing: a comparative study. BioRxiv.






