From kilobases to "whales": a short history of ultra-long reads and high-throughput genome sequencing
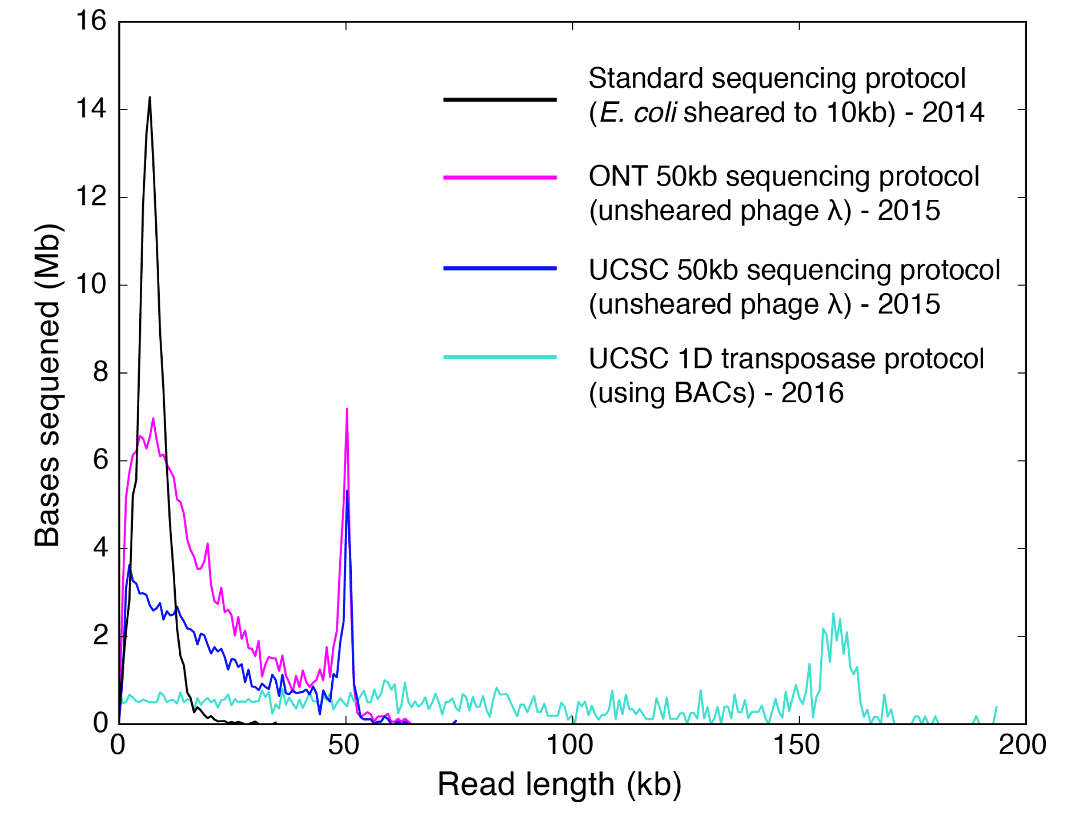
The evolution of long reads picked up speed when the MinION Access Programme began in 2014. Right away from the Nanopore burn-in experiments, 10 kb reads became routine (10 kb because, as per recommendation, users would shear phage lambda DNA to 10 kb). From 2014 to 2016, several groups in the research community and within Oxford Nanopore Technologies developed methods that yielded systematic populations of long reads using the conventional ligation chemistry and early-access transposase chemistry (Figure 1). In 2016, Oxford Nanopore Technologies released the transposase-based rapid kits in early access. The chemistry was simpler and required less steps. Importantly, it got rid of SPRI-bead cleanups which were presumed to be a detriment to Nanopore read lengths. Applying the transposase-chemistry to BACs (bacterial artificial chromosomes), our group demonstrated that it was possible to get long reads where the peak of the read length distribution coincided with the size of the linearized BAC itself.
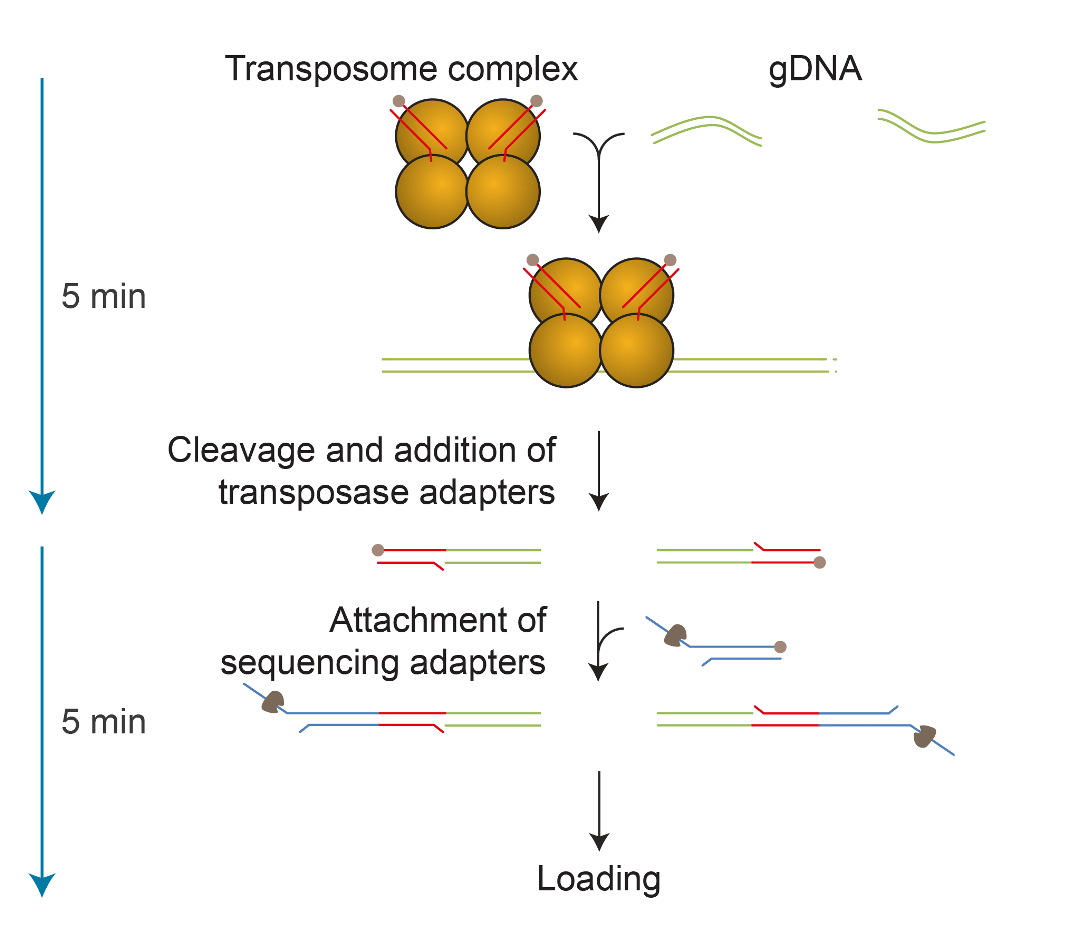
An update to this transposase kit was released in 2017, which brought the accuracy improvements to these long reads. The new kit was used for assembling the first human centromere at UC Santa Cruz1. In parallel, Josh Quick and Nick Loman used this kit for generating 100 kb+ reads as part of the Nanopore human genome project2. The new schematic for the rapid kit adaptation process is described in Figure 2.
The read lengths obtained by Josh and Nick were so unprecedented that a whole new term was coined for long reads. Reads exceeding 100 kb in length were termed “ultra-long” and reads longer than a megabase were termed “whales”. This was also the advent of whale watching, where users would sequence high-molecular weight DNA adapted using the rapid kit, and wait for the whales to show up over the duration of the experiment. The long reads kept showing up across several research groups, most notable of which was one of 2.3 Mb in length3, which held the record for a while. This record was recently overtaken by a 4.2 Mb read that was observed in an internal Oxford Nanopore Technologies run, announced on Twitter.
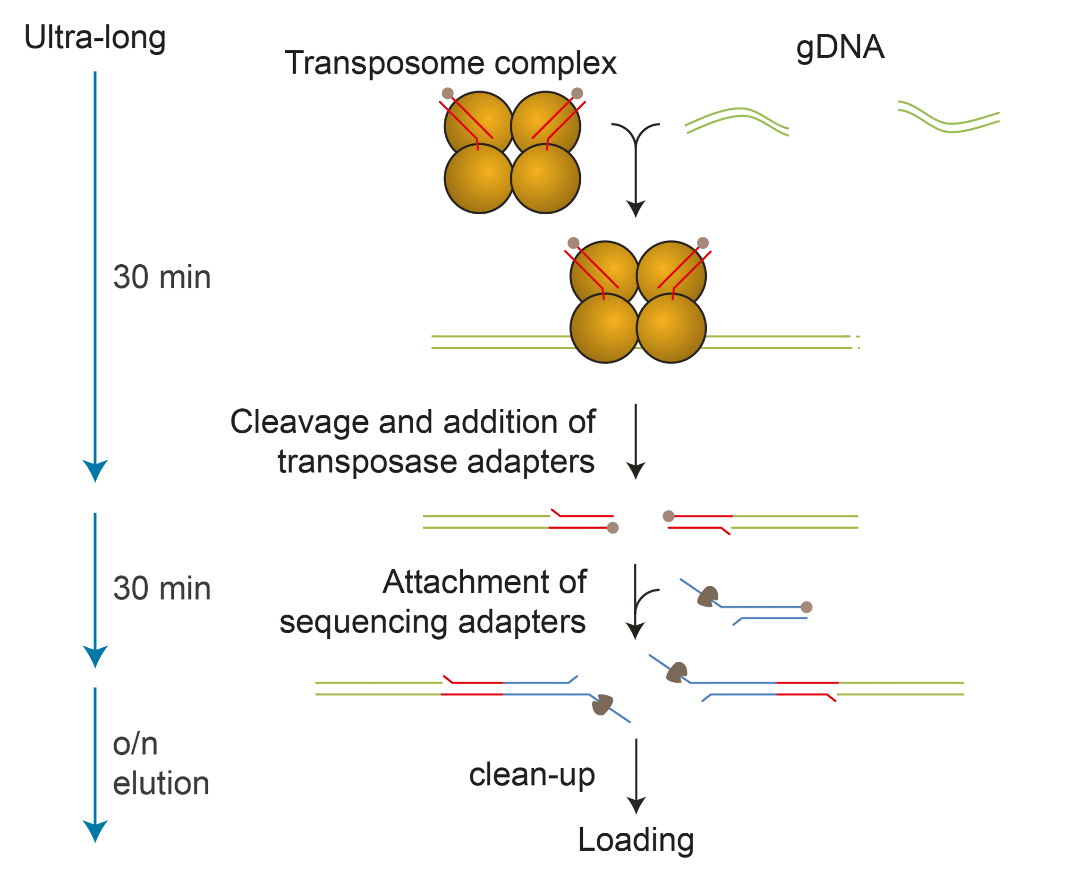
One of the limitations of this new protocol was that it did not scale well on to the PromethION. Several groups, both academic and industrial, have been developing methods for systematic, high-throughput, long-read sequencing using the PromethION. A recent advance to this process was the development of the ultra-long kit (SQK-ULK001) by Oxford Nanopore and Circulomics. Briefly, this kit uses Circulomics technology for high-molecular weight DNA extraction, library prep with rapid kit chemistry (Oxford Nanopore Technologies), and then library clean up (Figure 3). Its simple set up and easy-to-execute protocol works really well on the PromethION. Typically, this protocol includes a nuclease flush on the flow cell every 24 hours. This means that every PromethION Flow Cell receives three libraries of the same kind for maximizing throughput.
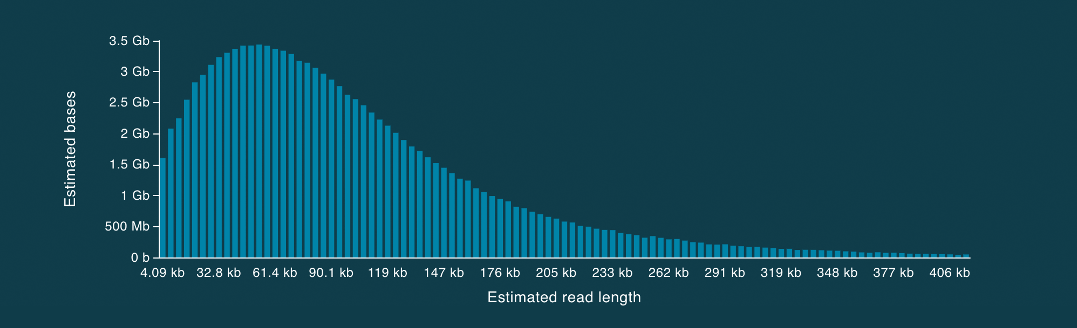
An example read length histogram using the SQK-ULK001 kit is shown in Figure 4. It is worth noting that there is a healthy distribution of long reads extending into hundreds of kilobases in length. Additionally, several whales are yielded by each sequencing experiment.
There are several applications of ultra-long nanopore reads. These include improvements to de novo assembly and phasing. We have recently shown that using these long reads, we can perform high-contiguity phasing4. Additionally, ultra-long reads help improve assembly contiguity. We have previously shown the impact of these reads on assembly contig NG50 using HG002 as a model. Briefly, a Shasta-based assembly of ligation-based long-read HG002 data5, basecalled using Guppy 3.6.0, yielded a 32.3 Mb contig NG50. In contrast, when we perform the assembly using 100 kb+ reads, the resulting contig NG50 was 88.3 Mb. We recently also performed a Shasta assembly for the CHM13 ultra-long dataset released by the Telomere-to-Telomere consortium, rebasecalled using Bonito 0.3.1, and noted a contig NG50 of 90.6 Mb. In this assembly, 36 of the chromosome arms were assembled in individual pieces.
The new improvements in basecaller models help not only with contiguity but also with accuracy. Using trio-binning on HG002 data generated at UCSC using Q20 early-access chemistry, we have observed haplotypes being assembled at accuracies exceeding Q39. Furthermore, we can now perform high-quality single nucleotide variant calling using nanopore reads that is on par with performance obtained using short-read technology.
These developments are contributing to several exciting genomics applications, such as a complete human genome6. Ultra-long reads are a significant player in the Human Pangenome Reference Consortium which will generate over 350 reference-grade, high-quality, reference genomes. Additionally, long reads are being used in large-scale population cohort sequencing projects such as NIH CARD, and Genomics England, among others. We anticipate significant strides in assembly quality, phasing, and structural variation inference as nanopore chemistry and basecalling software improves further. Examining DNA modification information over long range genomic contexts will facilitate biological discoveries. This will mark a new era for human genomics, and contribute towards realizing precision medicine.
- Jain, M. et al. Linear assembly of a human centromere on the Y chromosome. Nat. Biotechnol. 36, 321–323 (2018).
- Jain, M. et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 36, 338–345 (2018).
- Payne, A., Holmes, N., Rakyan, V. & Loose, M. BulkVis: a graphical viewer for Oxford nanopore bulk FAST5 files. Bioinformatics (2018) doi:10.1093/bioinformatics/bty841.
- Shafin, K. et al. Haplotype-aware variant calling enables high accuracy in nanopore long-reads using deep neural networks. bioRxiv 2021.03.04.433952 (2021) doi:10.1101/2021.03.04.433952.
- Shafin, K. et al. Nanopore sequencing and the Shasta toolkit enable efficient de novo assembly of eleven human genomes. Nat. Biotechnol. (2020) doi:10.1038/s41587-020-0503-6.
- Nurk, S. et al. The complete sequence of a human genome. bioRxiv 2021.05.26.445798 (2021) doi:10.1101/2021.05.26.445798.






