London Calling 2021: Day 3
Over 6000 delegates could settle in on Friday morning to partake in another day of talks, demonstrations and panel sessions on the latest cutting-edge science being carried out with nanopore sequencing. With a greater density of plenary talks, see below for a comprehensive outline of the day.
Welcome - Zoe McDougall
Zoe reminded us of the fact that at London Calling we're looking for ways to help foster collaboration between attending scientists, reminiscing back to the meetings of Crick and Watson, and Srinivasa Ramanujan and G.H. Hardy, and their desire to collaborate in-person. Off the back of this she hopes you'll be with us in London, in person, in 2022. She used Devin O'Rourke also as an example of how London Calling networking influenced the path his PhD took and where he finds himself now. Before introducing a video demonstrating the role nanopore sequencing has helped to play in the Covid-19 global pandemic, she poignantly touched on how SARS-CoV-2 had lead to 'one of the most astonishing examples of scientific collaboration'. It was then into the days talks.

Nanopore sequencing of cervical cancers uncovers novel genomic, epigenomic, and transcriptomic features associated with HPV integration events - Vanessa Porter
In sub-Saharan Africa cervical cancer is the second most prevalent cancer type and the number one cause of cancer mortality. The objective of Vanessa Porter's project is to identify the genomic and epigenomic correlates associated with human and viral gene dysregulation around HPV integration events using nanopore sequencing technology.
HPV genomic integration occurs near to cancer genes (e.g. TP53 and MYC) in the host genome, and often dis-regulates host gene expression. Using nanopore sequencing on two cohorts (one from Uganda and the other from the USA) Vanessa and her team are characterising these integration sites in HPV tumours. Long nanopore reads generated on the PromethION have permitted phasing of haplotypes and read-specific methylation calling. In addition to this they have used whole-transcriptome nanopore sequencing of HPV tumours, with their whole pipeline consisting of mapping, structural variant and CNV calling, de novo assembly, methylation, haplotype phasing, and fusion calling.
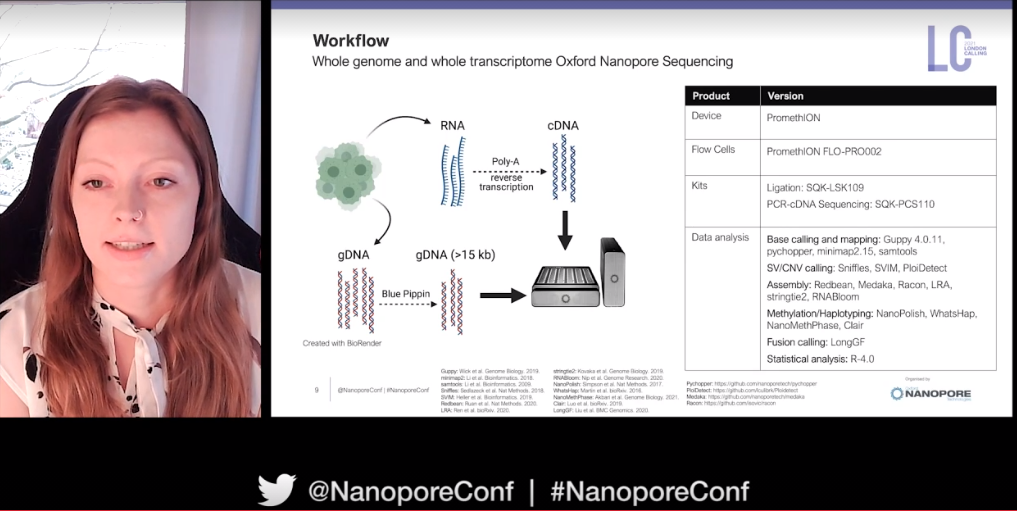
Vanessa has been able to identify either a single clonal integration event or multiple integrations in cervical tumour samples. Focusing on multi-integrated samples, one example demonstrated integration within at least three chromosomes whilst another complex integration event involved three genes on chromosome 12. Quite simply, 'this type of phenomenon would be difficult to find if not for long-read sequencing and our integration site probing technique’.
Switching to look at methylation, evidence suggested that integrations were able to contribute to demethylation of regulatory regions on the human DNA, and this was followed swiftly with a look at transcriptional activity from HPV integrations. Of 5 major versions of HPV polycistronic transcripts she identified, the second most highly expressed had not been described before to the best of her knowledge.
More great work is planned by Vanessa to sequence additional samples with nanopore technology, continuing investigations into the overall patterns of how HPV integration in different HPV types affects the structure and regulation of cervical cancer genomes.
Innovation with ultra-long reads - Panel plenary
While three can sometimes be considered a crowd, it's also the number at which something becomes scientific. Therefore, the panel plenary session on ultra-long reads featuring three ultra-interesting talks had the keen attention of all the attendees.

First up was Matthew Brian Couger of Brigham and Women's hospital, USA, talking about Ultra-long nanopore sequencing for assembly and scaffolding of sex chromosomes in the creeping vole. The biological puzzle surrounding this animal's novel sex chromosome system started in the 1960s. Fascinated by this Brian's team, lead by Polly Campbell at UC Riverside, began to explore it using genomic sequencing.
Short-read sequencing proposed a possible mechanism, however the placement and order of Y sex chromosome genes was still unclear, and with even long reads failing to adequately resolve the sex chromosomes they had to turn to ultra-long reads to cover the repetitive sequences and bridge their contig gaps. With a month to complete the project, Matthew highlighted how this was a very real-world example of ‘how you can use really long reads in a project that’s not…taking you another 6 months to do all this work’.
Using the SQK-ULK001 kit they generated two PromethION flow cells worth of data with a read N50 of 91kb. This ultra-long data aligned well to the genome, allowing them to connect unassembled regions of the repeat-rich sex chromosomes. The position of genes relative to each other, both within a chromosome, and between the paternal X and maternal X chromosomes, was revealed.
Following Matt was Jillian Hammond of the Garvan Institute, Australia, with her talk on Ultra-long reads from Australian reptiles. Starting with the bearded dragon and the shingleback lizard, a specific aim of hers was to discriminate between the sex chromosomes, which contribute to the novel mechanism whereby the sex of the species is determined by both the genotype and the environment, particularly the temperature. Typically a ZZ genotype becomes a male and a ZW genotype becomes a female, but at higher egg incubation temperatures this is overridden and both become female. The mechanism for this has been obscured by the highly repetitive and homologous Z/W chromosomes, which ultra-long reads can help to elucidate.
Jillian's second project involves two highly venomous sea snakes, but this time aiming for full reference genomes to provide insight into how they have adapted to the specialised marine environment; one such way being their ability to take oxygen in through their skin and expel nitrogen and carbon dioxide. The genes and underlying mechanisms for this phenotype remain unclear.
Now the ultra-long sequencing kit has become available she sees over a third of her reads exceeding 100kb, with a significant number exceeding 500kb. Her work is still in its infancy but she has draft genome assemblies already available, and some polishing of genomes already completed. She also gave a nod towards the transcriptome for these species, intending to perform cDNA sequencing for annotation and elucidation of the mechanisms which make these animals so unique.
The final member of the panel was Roham Razhagi from Johns Hopkins University, USA, shedding light on the long-range interaction of the human epigenome using ultra-long nanopore sequencing. While gene promoters direct RNA polymerase to a transcription start site, enhancers are gene expression regulatory regions which can be up to 1 Mb away from the transcription start site. The methylation of these regions has a nuanced effect on gene silencing, and Roham wished to 1) quantitatively look at interactions and methylation status at the single-cell level, and 2) distinguish between allele specific interactions. He believes ultra-long '‘nanopore sequencing could address both of these concerns'.
Typically generating 'more than 100 Gb of data with an N50 of around 100 kb on a single PromethION flow cell’, he performed his analysis using a putative driver of cancer progression, TBX18, as an example. The utility of ultra-long nanopore reads let him look at the discrepancy in enhancer and promoter methylation between the two haplotypes culminating in their respective gene silencing and gene activation.
Using the gene CCN1 (CYR61)Roham then demonstrated capturing more than one interaction between the promoter and enhancers. The generation of a a novel looping interaction model between this gene and two downstream enhancers was possible due to ultra-long reads showing heterogenous methylation status in these two regions. He had ‘not been able to capture this information with other methods’ and is optimistic that this will help with discovery of novel biology.
A discussion conducted by Oxford Nanopore's Rachel Rubinstein followed, with noticeable responses including the answer to the question "What is the longest read you have observed with the ultra-long kit?" being 'around 4Mb', '2.5 Mb was probably the longest we got' and 'our longest was 2.2 Mb'.
Long-read metagenomic sequencing of faecal samples to study convergent dietary adaptation in ant-eating mammals - Sophie Teullet
Myrmecophagy. Not a word you may be familiar with, but it describes a feeding behaviour focused around ingesting ants or termites. The myrmecophagous phenotype also appears to be a result of convergent evolution, with aardvark, ground pangolin and the southern aardwolves all displaying this behaviour. Sophie Teullet wanted to uncover how each of these species adapted to their novel diet, with a focus on metagenomics and the gut microbiome.
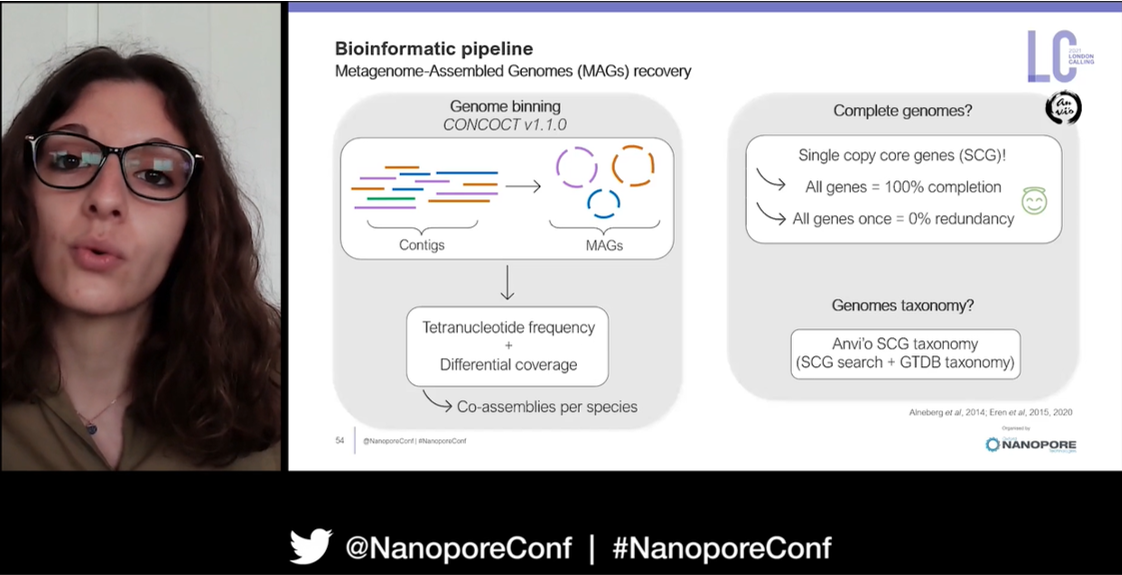
To begin her research in French Guiana and South Africa she had to collect faecal samples either from roadkill, or by following animals back to their burrows. DNA was then extracted from this and long read sequencing conducted on a MinION Mk1C. Guppy was used for basecalling, Porechop for adapter removal, and MetaMaps for filtering reads from different species.
Taxonomic profiling identified the main bacterial phyla in the gut metagenomes with Bacteroidetes, Firmicutes, and Proteobacteria were all present. Specifically for chitin degradation, it appeared the Bacteroidetes Chitinophaga were conferring an advantage on the animals as they were found in all metagenome samples. Sophie then went on to investigate the genomes of these organisms using de novo assembly, with the hope of uncovering genes coding for chitinase proteins; specifically the GH18 genes which encode a family of enzymes that degrade chitin and related polysaccharides.
Evidence for this was discovered, and comparing data from the ground pangolin and a Malayan pangolin generated from a previous study, she found an overlap of several species all sharing chitinolytic genes. However when she directed her attention to the sequences presenting an active chitinolytic domain, it revealed that not all these were similar to known bacterial chitinases. Some for instance possessed a peptidoglycan-binding domain – whether it can bind chitin remains to be determined.
Sophie concluded with her next steps, which will help to shed light on whether bacterial species carry the same genes between the different myrmecophagous species and more generally understand if the same mechanisms are involved in the adaption to this diet. She also wishes to conduct sequencing with the MinION Mk1C in-field.
Leveraging adaptive sampling of environmental DNA for monitoring the critically endangered kākāpō - Lara Urban
Continuing the theme of animals and samples derived from the wild, Lara Urban of the Univeristy of Otago told us about the kākāpō. This is a critically endangered, flightless, nocturnal parrot native to New Zealand. Only 204 of them are alive today. It's vital therefore to be able to monitor them in their own ecosystem.

Lara's team setup an eDNA approach for monitoring the kākāpō population, which is almost exclusively found on remote predator-free islands. DNA extraction from soil samples permitted them to amplify the 12S ribosomal RNA region, and they found high abundance of kākāpō specific 12S genes from their sampling sites. This allowed them to identify species, but to detect individuals they required a whole genoes sequencing approach.
Using the GridION and adaptive sampling, Lara aimed to enrich the 1 Gbp kākāpō genome. Variant calling and phasing identified 26.4 thousand SNPs and 476 haplotypes, and combining this with more data from t kākāpō125+ consortium they proceeded to use SNPs and haplotypes to arrive at their 'eureka moment' and for the first time ever, identify individuals in a population from eDNA - even allowing them to give specific names to who they found.
Lara finished up by detailing the biodiversity monitoring project she setup for recovery of the Takahē, using long nanopore reads to generate a reference genome, and thanking her team members Jo Stanton and Miles Benton.
Effective characterization of T-DNA insertion lines through nanopore sequencing - Boas Pucker
Before CRISPR/Cas9, knock-out studies to investigate gene function had to be achieved with different means. In the plant model organism Arabidopsis thaliana this was conducted using the soil bacterium Agrobacterium tumefaciens, which injects ‘transfer DNA’ (T-DNA) into plant cells, which is then randomly integrated into the plant’s genome and can disrupt gene function. Boas Pucker of the University of Cambridge, UK, highlighted how this has been used to generate over 700,000 T-DNA insertion lines.
The project GABI-Kat aims to identify the locations of these insertions, however the classical method for this can sometimes fail. Long nanopore sequencing reads were used by Boas and his team to investigate these failures, with mechanisms such as T-DNA fragmentation, or multiple insertions being identified.
Going a step further, they were able to characterise insertions associated with structural variants, including cases were insertions had connected different chromosomes. It was also possible to find duplications by analysing their sequencing coverage across the genome.
In the process of this they generated ~700x coverage of the A. thaliana genome, which inspired them to produce a new de novo genome assembly once reads containing T-DNAs, SVs, and <100kb had been excluded. This new assembly was able to help resolve a T-DNA insertion in a peri-centromeric region which contained gaps and misassemblies in the TAIR reference genome.
Deciphering the role of splicing factor mutations in hematopoiesis via integration of long-read sequencing in single-cell multiomics - Dan Landau
New York Genome Centre's Dan Landau opened his plenary talk by explaining how tumour evolution is a fundamental process in cancer biology, associated with the development of a more aggressive phenotype over evolutionary time. Dan's lab aim to apply multi-omics solutions to capture these changes at the level of the single cell.
Starting with GoT (Genotyping of Transcriptomes), which involves single-cell RNA sequencing plus somatic mutation analysis, they were able to observe the link between mutation, cell phenotype, and mutant fitness in cell differentiation. Nanopore long reads were introduced when they looked at the introduction of new subclones over time, with complex clonal structures (for example in the gene SF3B1) demonstrating mutations far from transcript ends which could not be genotyped with short reads.
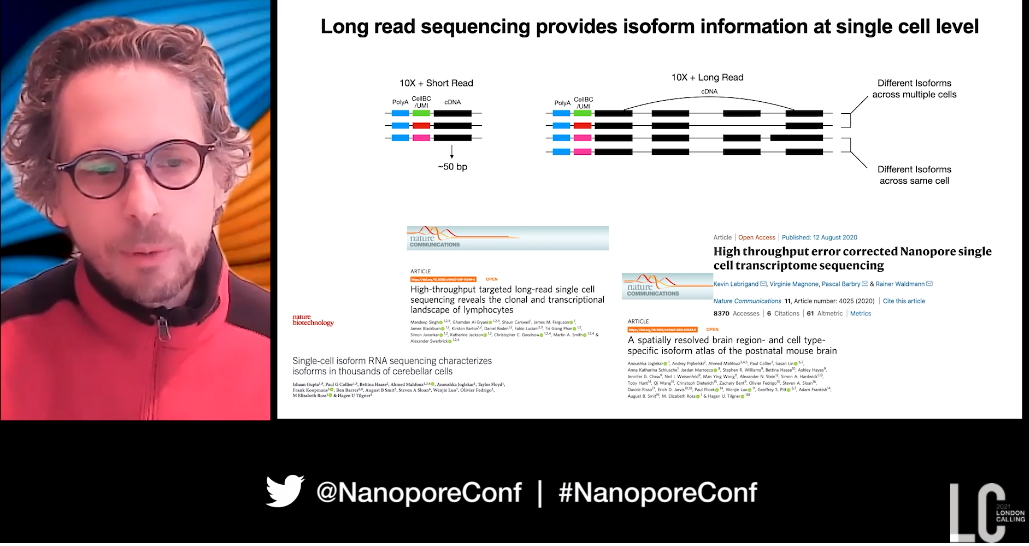
Dan then moved onto their work linking splicing factor mutations to cell phenotypes, and they knew they could use nanopore sequencing to reveal isoform information at the single-cell level from full-length cDNA sequencing. Nanopore reads from the PromethION gave a ‘massive increase in the number of junctions that we were able to capture per cell’.
Interestingly, Dan closed by pointing out how single-cell data from many tissue types, across numerous studies, have shown that ‘we are all mosaics’ and all cells in a body are not genetically identical. With aging, normal tissue is displaced by clonal outgrowths and before there are any obvious morphological or functional abnormalities, cell-type specific mis-splicing events can be seen. Observations like this will help as part of a larger multi-omics effort to tackle these clonal mosaics – to use novel technologies to go beyond genotyping, and link mutations to function.
Coronavirus genomic surveillance: Where have we been, where are we going next? - Nick Loman
The initial identification of a cluster of cases of fatal respiratory illness In Wuhan, China in 2019 is where Nick Loman of the Universty of Birmingham, UK began his talk. A few weeks later metagenomic sequencing revealed an association with the novel coronavirus that became known as SARS-CoV-2, and on January 10th Yong-Zhen Zhang (Fudan University, China) shared an early genome sequence for the pathogen. This early work set the stage for the data sharing which would be a hallmark of the Covid-19 pandemic.
Following this the work of Josh Quick helped the ARTIC tiling protocol for SARS-CoV-2 come to fruition, with the cost of sequencing on nanopore devices now at under £20/sample. This has been coupled with the ARTIC pipeline, and together by late January 2020 an end-to-end system was available for the sequencing of SARS-CoV-2.
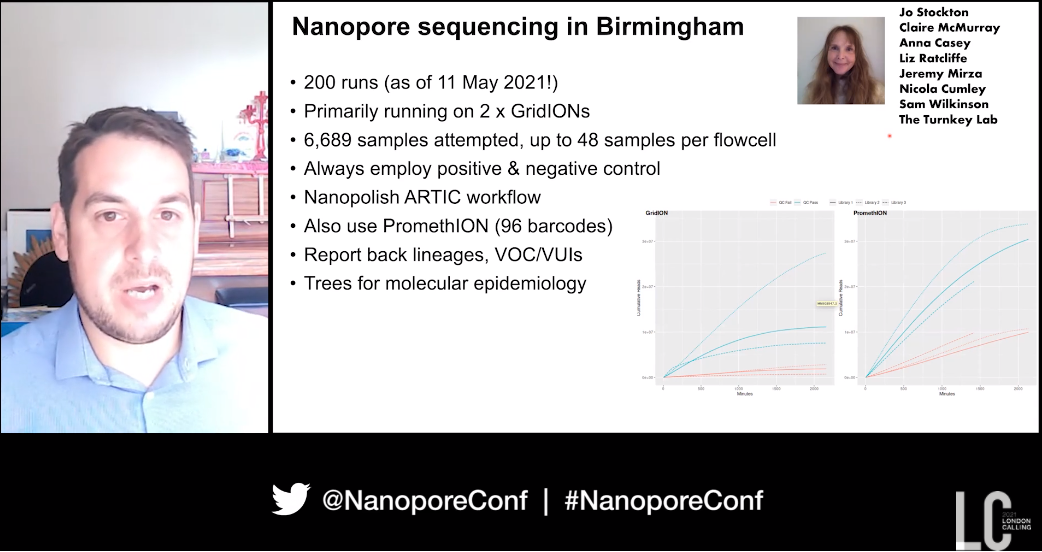
Nick went on to cover the creation of the COVID-19 Genomics UK Consortium (COG-UK), which from the outset decided that a “federated, distributed model" was essential. Early in the pandemic COG-UK linked sequencing data with travel data to estimate how COVID-19 came to the UK, with over 1300 independent introductions of the virus from mainland Europe into the UK seen at the start of March.
COG-UK also illuminated the 'shift in gears' of the virus in December 2020 with the appearance of the B.1.1.7 Kent variant and how this compared to the global situation; sharing of data and knowledge also identified similar mutations with the South African variant. Genomic modelling revealed the speed at which the B.1.1.7 variant outcompeted others each time it was introduced to a new part of the UK, showing an estimated 30-70% increased transmissibility.
Nick took pains to stress that the more the virus is allowed to spread, the more chances it has to evolve. If this occurs in areas without sufficient public health measures in place then it is more likely SARS-CoV-2 will evolve increased transmissibility or immune evasion ‘and that is what we, as a community, need to try and stop now’. The current situation in India was given as an example of that.
Nick finished his talk by highlighting examples of the use of the ARTIC protocol and nanopore sequencing method of SARS-CoV-2 genome analysis worldwide. Now, Nick said, it needs to be optimised and utilised further, and more funding is needed. Stressing that ‘nobody is safe until we’re all safe’, he concluded that ‘we need to establish that principle of real-time data sharing worldwide, because everything is connected and we’re all in this together’.






