Updated method for cell-free DNA (cfDNA) methylation profiling

Requirements
Updated method for cell-free DNA (cfDNA) methylation profiling
FOR RESEARCH USE ONLY.
Contents
Introduction
Methods and results
Recommendations
References
Change log
Introduction
Analysis of cell-free (cf)DNA methylation can be used for a range of diagnostics, including cancer detection and tissue-of-original analysis, and is an actively developing and emerging application. cfDNA is predominantly circulating within the blood as multiples of one or more nucleosome lengths, which results in a characteristic length profile that corresponds to fixed nucleosome positioning along the DNA. We and others (Jiang et al. 2020) have observed a sequencing platform independent, marked loss of methylation signal along cfDNA reads at regions corresponding to internucleosomal regions, and a sudden drop in the last ~30 base pairs, that can be attributed to library end preparation (Figure 1). Here, we demonstrate an updated DNA end-prep method using NEBNext FFPE DNA Repair v2 Module to better retain methylation in cfDNA reads, provide bioinformatic options to omit methylation information at specific read positions, and characterise the loss of methylation for the three most common cfDNA nucleosome lengths.
We have developed new updated methods that describe the methodology and analysis steps in detail:
- Ligation sequencing V14 — Human cfDNA singleplex (SQK-LSK114)
- Ligation sequencing V14 — Human cfDNA multiplex (SQK-NBD114.24)
The methods above replace our legacy human blood cell-free DNA (cfDNA) protocol.
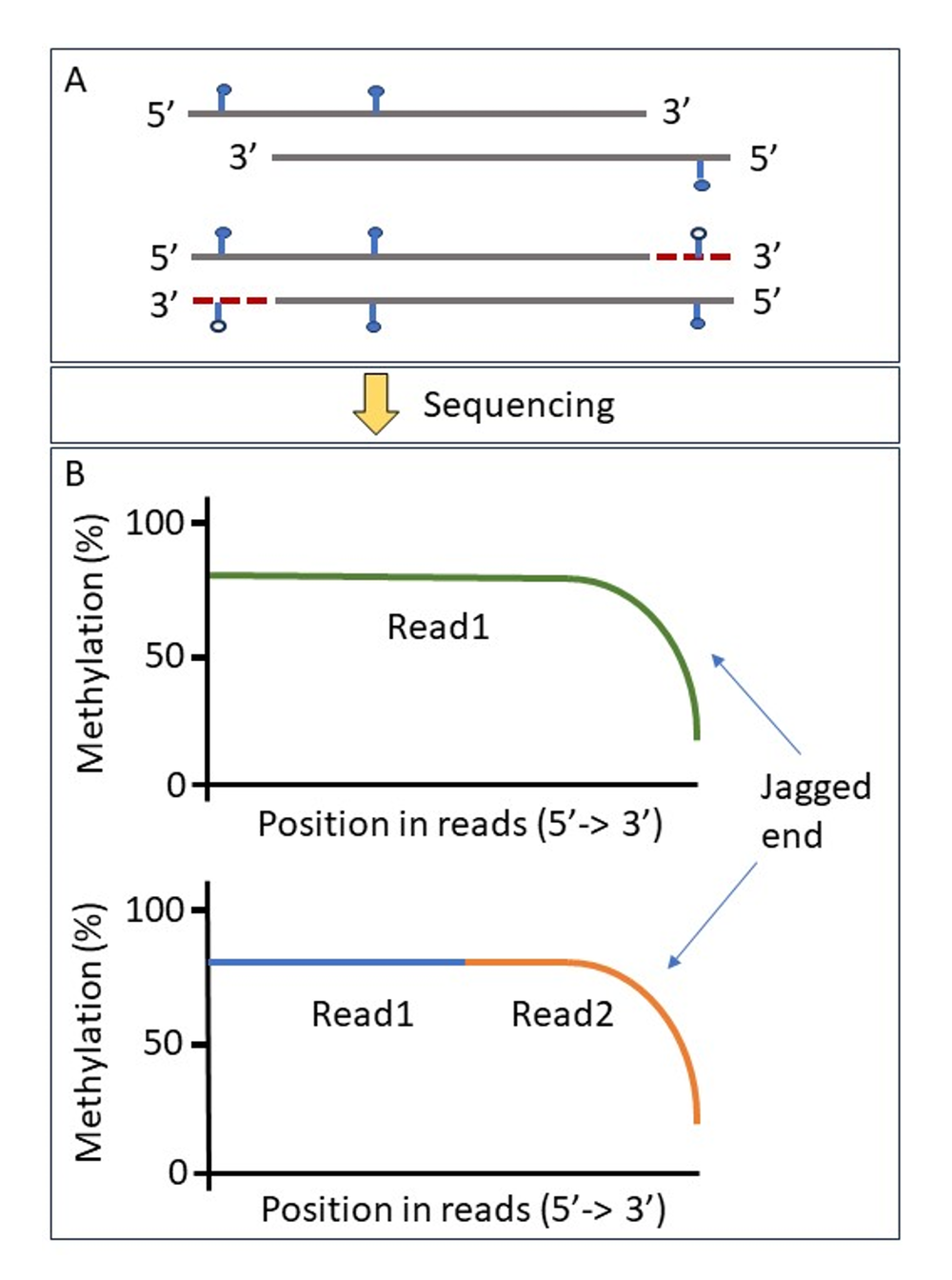
Figure 1. Example of mechanism of methylation loss of jagged end DNA. A) Filled blue lollipop sticks represent methylation and unfilled represent unmethylated. Red dashed line represents the filling in of complementary sequence. cfDNA double-stranded molecules with 5’ protruding ends (jagged ends) are filled in by unmethylated nucleotides during sequencing library preparation to produce blunt-ended molecules (Jiang et al). B) The resulting former jagged double-stranded single-stranded molecules would present with loss of methylation at the 3’ end of the molecules and lead to an increase in false negative unmethylated bases. This is evident when plotting the proportion of methylation based upon read base position from 5’ to 3’ where a decline in percentage methylation is observed across the data set. This is true for all sequencing types including Oxford Nanopore reads (green) and bisulphite sequencing (blue/orange).
Methods and results
We tested different library preparation conditions using a combination of End Prep modules (NEBNext Ultra™ II End Repair/dA-Tailing Module, NEBNext End Repair Module + NEBNext dA-Tailing Module, NEBNext FFPE DNA Repair Mix, and NEBNext FFPE DNA Repair v2 Module), with varied incubation and inactivation times. We then basecalled the data with Dorado high accuracy (HAC) model calling 5mCG and 5hmCG modified bases, then mapped against the human genome using minimap2, and extracted genomic coordinates for methylation using modkit.
We found that the FFPE DNA Repair v2 Module gives the best results, with almost complete recovery of lost methylation within the cfDNA fragments, thereby improving methylation detection especially for multi-nucleosome fragments (Figure 2). Impact on the methylation retainment at the end of cfDNA reads was modest. Libraries generated with this updated protocol retained the same high raw and aligned read output as our earlier method.
The updated protocol improved concordance with a methylation ‘truth set’. This set was generated using bisulphite sequencing, filtered for high-confidence methylated regions (regions with 20X coverage and 99% methylation confidence score) in an HG002 cell line. 91.0% of these sites were classified as methylated in cell-free samples using the current protocol; this increased to 96.0% with FFPE DNA Repair v2 Module, demonstrating that this protocol substantially mitigates methylation loss during cfDNA library preparation. Informatic trimming of 27nt off the ends of reads further increased this score to 97.4%. Due to the difference in source material, 100% capture of all 'truth set' methylation sites is not expected.
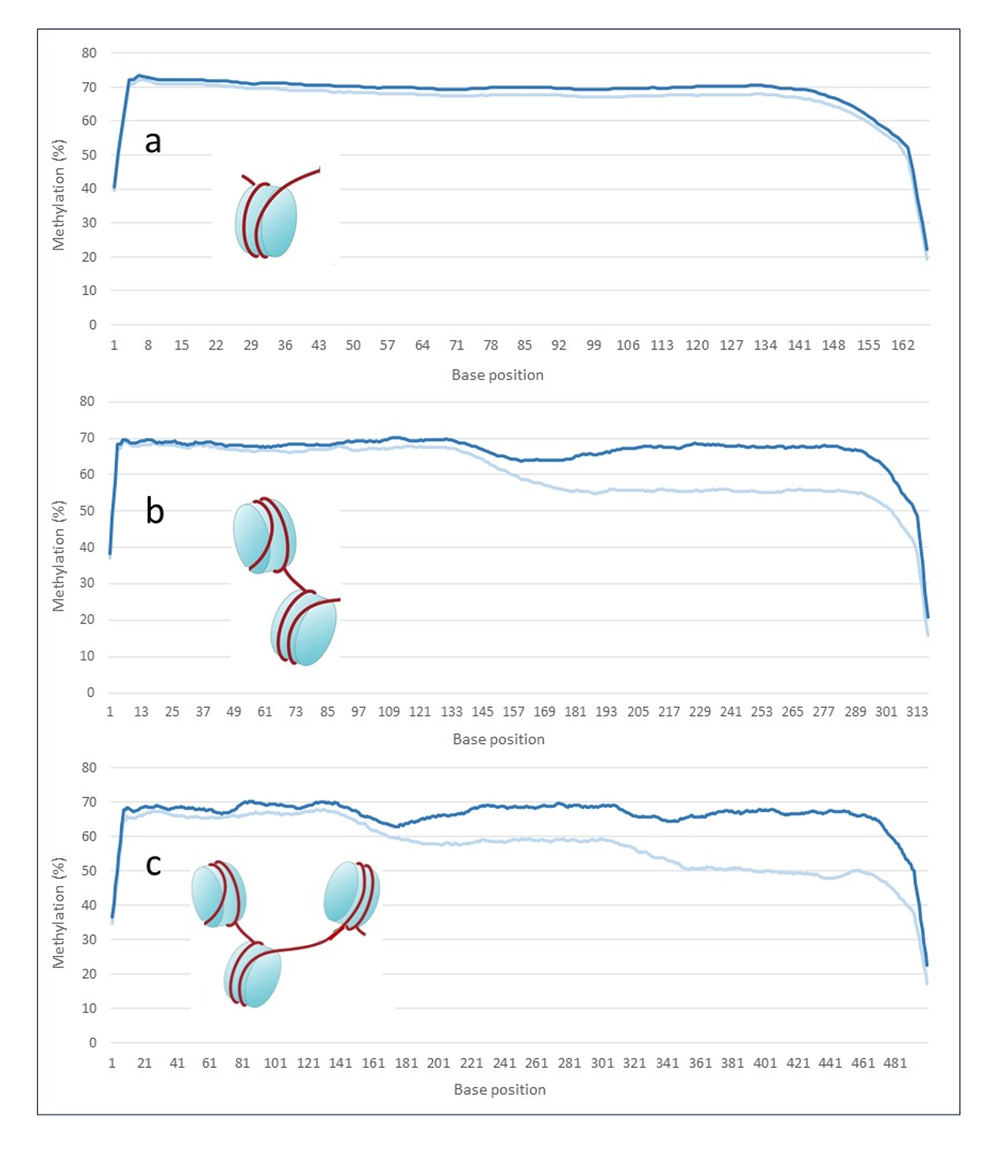
Figure 2. Average methylation frequencies for old method kit (light blue) versus the new recommendation using DNA Repair v2 Module (dark blue) (3x reps per treatment) across the length of the reads. Methylation along the read for reads falling within (a) single nucleosome of 167bp length, (b) double nucleosome of 317 bp length, and (c) triple nucleosome peak of 500 bp read length. The new protocol retains the methylation signal along the cfDNA reads.
Recommendations
To get the greatest methylation retainment and data output, follow our updated Ligation sequencing V14 — Human cfDNA singleplex (SQK-LSK114) protocol. This uses NEBNext Ultra™ II End Repair/dA-Tailing Module with NEBNext FFPE DNA Repair v2 Module and optimised incubation times. Ensure you are using the flow cell light shield when sequencing and take care to avoid light exposure to the flow cell during runtime to increase output. Basecall the data with the Dorado HAC basecalling model using the latest MinKNOW version or the latest standalone basecaller, with read trimming and splitting enabled. Align the reads to the genome of interest and filter for uniquely aligned reads with mapping quality above 10. Modkit adjust-mods can be used to remove methylation tags from the ends of the reads and produce an updated bam file. Downstream applications may use the resulting .bam for further analysis.
A multiplexing version of this protocol is also available to sequence 12 samples on a single flow cell: Ligation sequencing V14 — Human cfDNA multiplex (SQK-NBD114.24).
References
Jiang P, et al. Detection and characterization of jagged ends of double-stranded DNA in plasma. Genome Res. 30(8):1144-1153 (Aug 2020). 10.1101/gr.261396.120.
Change log
| Version | Change |
|---|---|
| v2, May 2024 | Updates including new protocol links |
| v1, Nov 2023 | Initial publication |






