Nanopore-only assemblies for simple, cost-effective surveillance of antimicrobial resistance
- shared.published_on: November 17 2022
At the London School of Hygiene and Tropical Medicine, UK, Ebenezer Foster-Nyarko and colleagues harnessed the long-read capability of nanopore sequencing to generate nanopore-only genome assemblies of Klebsiella pneumoniae — a common intestinal bacterium capable of causing life-threatening infections1. Over recent decades, K. pneumoniae, along with many other pathogens, has rapidly acquired antimicrobial resistance (AMR), and the World Health Organization now recommends robust surveillance of AMR as part of their critical Global Action Plan2.
'Nanopore … sequencing has rich potential for genomic epidemiology and public health investigations of bacterial pathogens, particularly in low-resource settings and at the point of care, due to its portability and affordability'
Foster-Nyarko, E. et al. Microb. Genom. (2023)
AMR profiling with nanopore sequencing
In their publication, Foster-Nyarko et al.1 noted that AMR determinants cannot be accurately delineated using short-read sequencing technologies because the ‘fragmented genome assemblies ... cannot accurately resolve plasmids and other mobile genetic elements that drive the dissemination of AMR determinants’. They further commented that nanopore sequencing reads generate contiguous genome assemblies, as the ‘longer reads ... are capable of resolving structural variations, long repeat regions, and genomic copy-number alterations’, at ‘Iow capital cost’1.
Although hybrid genome assemblies, containing both short-read and long-read data, have been used to overcome the limitations of short reads, continuous improvements in nanopore chemistry and analysis software now enable the generation of highly contiguous, highly accurate genome assemblies using nanopore sequencing alone.
Generating high-quality genomes to detect AMR genes
Ebenezer generated genomic libraries using the Native Barcoding Kit, enabling multiplexing of up to 24 samples in a single run, before sequencing on the MinION. Genome sizes ranged from approximately 5.1 Mb to 6.2 Mb, with a GC content of 49.6% to 57.7%. Exploring both time and accuracy in AMR determination, three different real-time basecalling models were applied: fast; high accuracy (HAC); and super accuracy (SUP).
A total of 270 acquired AMR genes were present across 54 reference genome assemblies, and the majority were correctly identified using SUP basecalling with Medaka polishing (95.8%). Mutations were identified in the gyrA and parC genes (substitutions associated with fluoroquinolone resistance), in blaSHV (substitutions associated with extended spectrum β-lactamase activity), and in ompK35 and ompK36 (truncations associated with reduced susceptibility to β-lactams) (Figure 1). Colistin resistance is associated with truncations in pmrB and mgrB — these genes were intact in all reference assemblies.
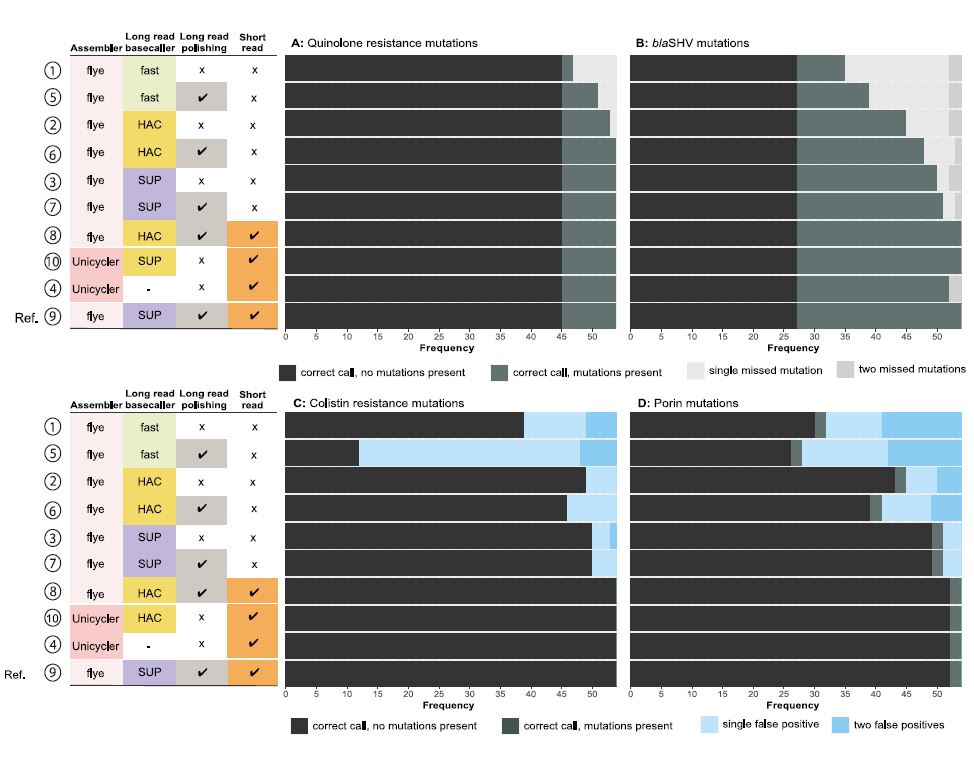
Figure 1: Detection of AMR-associated mutations. Each panel summarises the accuracy of genotyping across 54 samples for each assembly methodology, compared with the reference assembly. A) Detection of fluoroquinolone resistance associated mutations. B) Detection of mutations in blaSHV. C) Detection of colistin resistance-associated mutations. D) Detection of mutations in outer membrane porins which are associated with carbapenem susceptibility. (Image kindly provided by Dr Ebenezer Foster-Nyarko, London School of Hygiene and Tropical Medicine, UK).
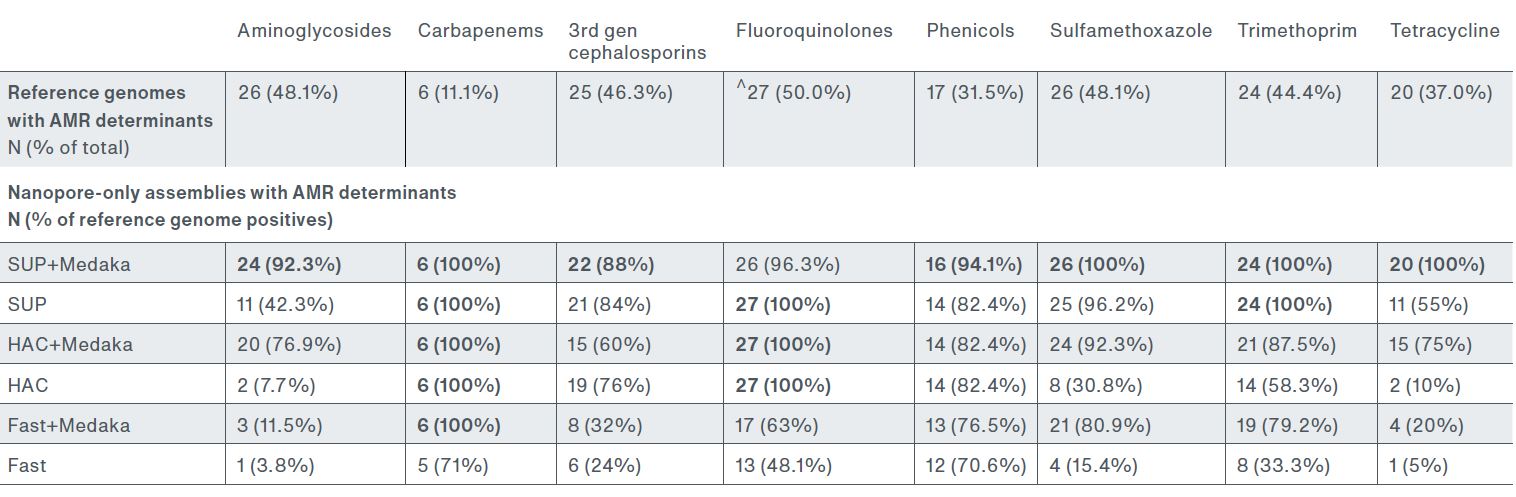
Table 1: Accuracy of identifying AMR based on the detection of AMR determinants in the genome assembly. Data provided by Dr Ebenezer Foster-Nyarko, London School of Hygiene and Tropical Medicine, UK.
‘There is ... now an unparalleled opportunity to harness Oxford Nanopore sequencing for genomic epidemiology and surveillance of bacterial pathogens, which is particularly attractive for AMR associated pathogens such as K. pneumoniae’
Foster-Nyarko, E. et al. Microb. Genom. (2023)
The presence of AMR-associated mutations is often used to predict drug resistance. In this research study, assemblies basecalled with SUP, assembled with Flye, and polished with Medaka identified ‘88% – 100% of genomes with AMR determinants ... across the various drug classes’ (Table 1)1.
Foster-Nyarko, E. et al. Nanopore-only assemblies for genomic surveillance of the global priority drug-resistant pathogen, Klebsiella pneumoniae. bioRxiv 498322 (2022).
Global action plan on antimicrobial resistance. World Health Organization. 2016. Available at: https://www.who.int/publications/i/item/9789241509763 [Accessed: 12 October 2022]






