Comprehensive characterisation of repeat expansions in neurodegenerative diseases with long nanopore reads

- shared.published_on: February 2 2022
Approximately 500,000 tandem repeats (TRs) have been identified in the human genome. These TRs are highly mutable with a notable propensity to expand1. It is these expanded repeats that are implicated in many neurological diseases, including amyotrophic lateral sclerosis and frontotemporal dementia2. Yet, despite their obvious clinical significance, they remain poorly understood; due to their repetitive nature, high GC content, and length, these expansions are refractory to PCR amplification and often cannot be spanned by short reads. For these reasons, repeat expansions are generally precluded from base-level resolution by sequence analysis based on short-read technologies.
Despite the technical challenges faced in characterising repeat expansions, research has demonstrated that there is a negative correlation between repeat length and the age of disease onset as well as symptom severity3. Both methylation status and base composition of the repeat have also been shown to alter disease phenotype. These observations suggest that to comprehensively characterise repeat expansions and understand their role in disease, the repeat length, methylation status, and base composition need to be determined.
'We achieved high single-read accuracy for both TR length and sequence, which opens novel avenues in TR research'
Roeck, A.D. et al. Genome Biol. (2019)
Due to the limited capacity of other technologies to resolve repeat expansions, Sleegers and colleagues based at the VIB Center for Molecular Neurology, Belgium, used long reads produced by Oxford Nanopore's PromethION device to investigate the role of the ABCA7 repeat expansion in Alzheimer's disease4. The group sequenced genomic DNA from 11 individuals who previously had their ABCA7 variable number tandem repeat (VNTR) allele size estimated using Southern blotting. For their analysis they developed NanoSatellite — a tool to determine repeat length and composition from the raw nanopore squiggle data (Figure 1). Using this workflow, they resolved 'all ABCA7 VNTR lengths, including expansions', which allowed them to obtain both 'high-quality TR length and sequence determination'.
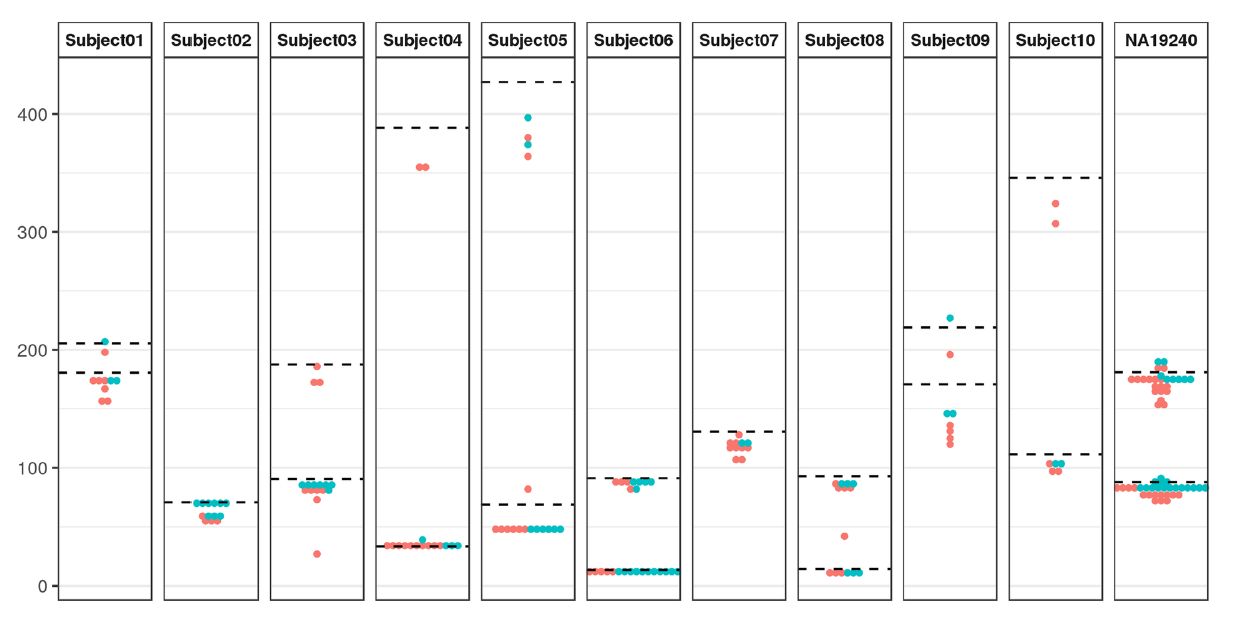
Figure 1: NanoSatellite ABCA7 VNTR length estimates for all individuals. The number of tandem repeat units (displayed on the y axis) per positive strand (red) or negative strand (blue) is shown in comparison to the lengths estimated via Southern blotting (dashed black lines). Image modified from De Roeck et al. 20194.
Franz-Josef Müller's team, at the Max Planck Institute for Molecular Genetics in Berlin, leveraged long nanopore reads to characterise amyotrophic lateral sclerosis causative C9ORF72 repeat expansions2. DNA was obtained from clinical research samples harbouring the C9ORF72 repeat; they then enriched for the C9ORF72 region using Cas9-mediated targeted enrichment, and sequenced samples on a single MinION Flow Cell. The workflow enabled an ~8-fold enrichment of C9ORF72 compared to non-targeted whole-genome sequencing. For their analysis, they developed the tool STRique, which, akin to NanoSatellite, analyses raw nanopore squiggle data, and after further alignment steps, accurately quantifies the number of repeats within an expansion.
Furthermore, as this method of enrichment also preserves epigenetic modifications, the team explored the methylation content of the repeat — something which would not have been possible if an amplification-based method had been used. Overall, Giesselmann and colleagues demonstrated the value of nanopore sequencing, which enabled 'precise quantification of repeat numbers in conjunction with the determination of CpG methylation states in the repeat expansion'2.
Fazal, S. et al. Large scale in silico characterization of repeat expansion variation in human genomes. Sci. Data 7(1):294 (2020). DOI: https://doi.org/10.1038/s41597-020-00633-9
Giesselmann, P. et al. Analysis of short tandem repeat expansions and their methylation state with nanopore sequencing. Nat. Biotechnol. 37(12): 1478–1481 (2019). DOI: https://doi.org/10.1038/s41587-019-0293-x
Paulson, H. Repeat expansion diseases. Handb. Clin. Neurol. 147:105–123 (2018). DOI: https://doi.org/10.1016/b978-0-444-63233-3.00009-9
De Roeck, A. et al. NanoSatellite: accurate characterization of expanded tandem repeat length and sequence through whole genome long-read sequencing on PromethION. Genome Biol. 20(1):239 (2019). DOI: https://doi.org/10.1186/s13059-019-1856-3






