Nanopore Community Meeting 2018 - Day 2 writeup

Plenary: Michael Schatz
Opening day 2 of the Nanopore Community Meeting 2018, Michael Schatz, Bloomberg Distinguished Associate Professor of Computer Science and Biology at Johns Hopkins University, described his group’s ground-breaking research characterising the structural variant landscape in tomato genomes. With an annual production of over 175 million tonnes and a value of $85B, the tomato is one of the most valuable crops in the world. They are also an important model plant system, exhibiting extensive variation across over 15,000 known varieties – providing a model for studying fruiting, taste and the important Solanaceae family, which includes potato and pepper.

Published in May 2012, the tomato reference genome was an international collaboration requiring years of effort costing millions of dollars. Michael noted that this has proved an invaluable resource for thousands of studies (including his own) and has delivered candidate SNPs for many traits.
According to Michael: ‘Short-read sequencing has proven valuable for single nucleotide polymorphism (SNP) discovery, but lacks power for more complex structural variants (SV)’. Recent research has highlighted that SVs play a major role in phenotypic variation, making their study increasingly important. Michael commented that long-read sequencing has the power to uncover this previously hidden variation; however, until recently, it had been too expensive to apply at scale. This all changed with the launch of the PromethION. Taking advantage of the facility for rapid and affordable long-read analysis of SV, Michael outlined his team’s involvement in a multicentre project to characterise SV landscapes in 100 diverse tomato genomes in just 100 days. The aim of the study is to elucidate the role of SV in natural variation, domestication and crop improvement. Over 900 tomato species have been sequenced to date with short reads, most at approximately 20-40x coverage. Michael described how they wanted to select the most diverse varieties to cover the maximum amount of structural variation possible. Simply selecting varieties at random underrepresented the diversity, so the team wrote an algorithm that utilises the SNP information gleaned from the existing genome sequences to ensure the maximum genomic diversity in the study samples.
Their initial intention was to mix long- and short-read sequencing; however, this all changed based on successful test runs using the PromethION, which, in their hands, is currently yielding up to 109 Gb per flow cell. They are now running 12-16 samples per week allowing them to easily hit their target of 100 genomes in 100 days. The team use the Ligation Sequencing Kit, (LSK109) which offers both long reads and high yield. The current mean read length across all of the genomes sequenced to date is 10-20 kb; however, lengths up to 1.5 Mb have been obtained. They are now generating approximately 1 Tb of sequencing data per week, which Michael described as a ‘world changing phenomenon’. He also identified that such high-throughput sequencing capability creates new data management challenges, which they have addressed through increasing their data storage capacity and upgrading their data network.
Next, Michael discussed the two major strategies for SV analysis: alignment-based detection and assembly-based detection. Both offer unique advantages and challenges. Alignment-based analysis using their own aligner, NGMLR, provided significantly more accurate SV detection than BWA-MEM. Even small structural variants are enough to cause systematic alignment errors in short-read data; however, long reads allow sequencing of the entire structural variation. Also SVs are often localised with repetitive sequence, which precludes discovery using short reads.
For de novo assembly-based approaches, Canu worked well, delivering contig N50 sizes 10-fold better than the reference, but at the expense of speed – taking two weeks per assembly using 320 cores. The team are now exploring alternative, faster assembly options, including miniasm, wtdbg2 and cloud-enable pipelines. Michael commented that these are ‘reference quality assemblies with contig sizes up to 30 Mb’.
Michael also presented a novel method for fast and accurate reference-guided scaffolding called Ragoo. Using this tool, it is possible to generate almost complete chromosomes, in a significantly faster timeframe and with more accuracy than the popular salsa algorithm. Subsequent structural variation identification is achieved using their Assemblytics pipeline.
Initial results from the first 12 genomes sequenced in this project showed substantial variation between samples, with between 25,000-45,000 SVs each. Most of these SVs were insertions and deletions and, while the majority of variants are specific to each sample, a number of variants are shared by multiple samples – including some in all 12 samples.
Michael also shared data validating an 83 kb duplication spanning the ej2 gene that counteracts a negative epistatic interaction commonly found in crossed tomato plant lines. This has allowed the team to utilise CRISPR/cas9 approaches to overcome such negative epistatic interactions to improve fruit yields. According to Michael, ‘this is just first of many stories’ that he expects to come out of this study.
Summarising his talk, Michael reiterated that long-read nanopore sequencing on the PromethION has allowed the identification of thousands of variants previously missed using short-read sequencing. He also highlighted that nanopore sequencing allows the generation of genome assemblies ten times better than the original tomato reference genome within a couple of days and at a fraction of the cost. The high-throughput provided by PromethION has allowed the team to do this at a scale that has never been seen before, and already they have generated more reference quality assemblies than any other plant or animal species.
Michael has kindly made his presentation slides available to the research community.
Breakout: Raw signal analysis
Jared Simpson
Jared Simpson, Principal Investigator at the Ontario Institute for Cancer Research is well known amongst the nanopore community having developed nanopolish – one of the foremost tools for the analysis of nanopore data. As Jared explained, nanopolish is essentially a suite of tools for working with signal-level nanopore data and includes consensus calling, methylation detection, reference-based SNP calling and signal alignment. Jared kicked off his presentation by discussing the use of nanopolish for the detection of base modifications. He explained that this is a particularly exciting area as nanopore sequencing allows direct detection of methylated bases without the need for chemical treatment such as bisulfite conversion. Much of the early work was done in collaboration with Winston Timp, who developed the method in which training data is generated to inform the nanopolish calling. This proof of principle work was published in nature methods in 2017. More recently, as part of the nanopore human genome sequencing project the team used nanopolish to call methylated based with high concordance to bisulfite sequencing. Jared mentioned that a lot of research is now taking place in this area and pointed out some ongoing work studying imprinting. Nanopolish also allows the detection of other nucleotide analogues, including synthetic bases such as BrdU. This work is being done in collaboration with Conrad Nieduszynski at University of Oxford. They have shown that it is possible to detect BrdU in replicating yeast cells to identify origins of replication across the yeast chromosome.
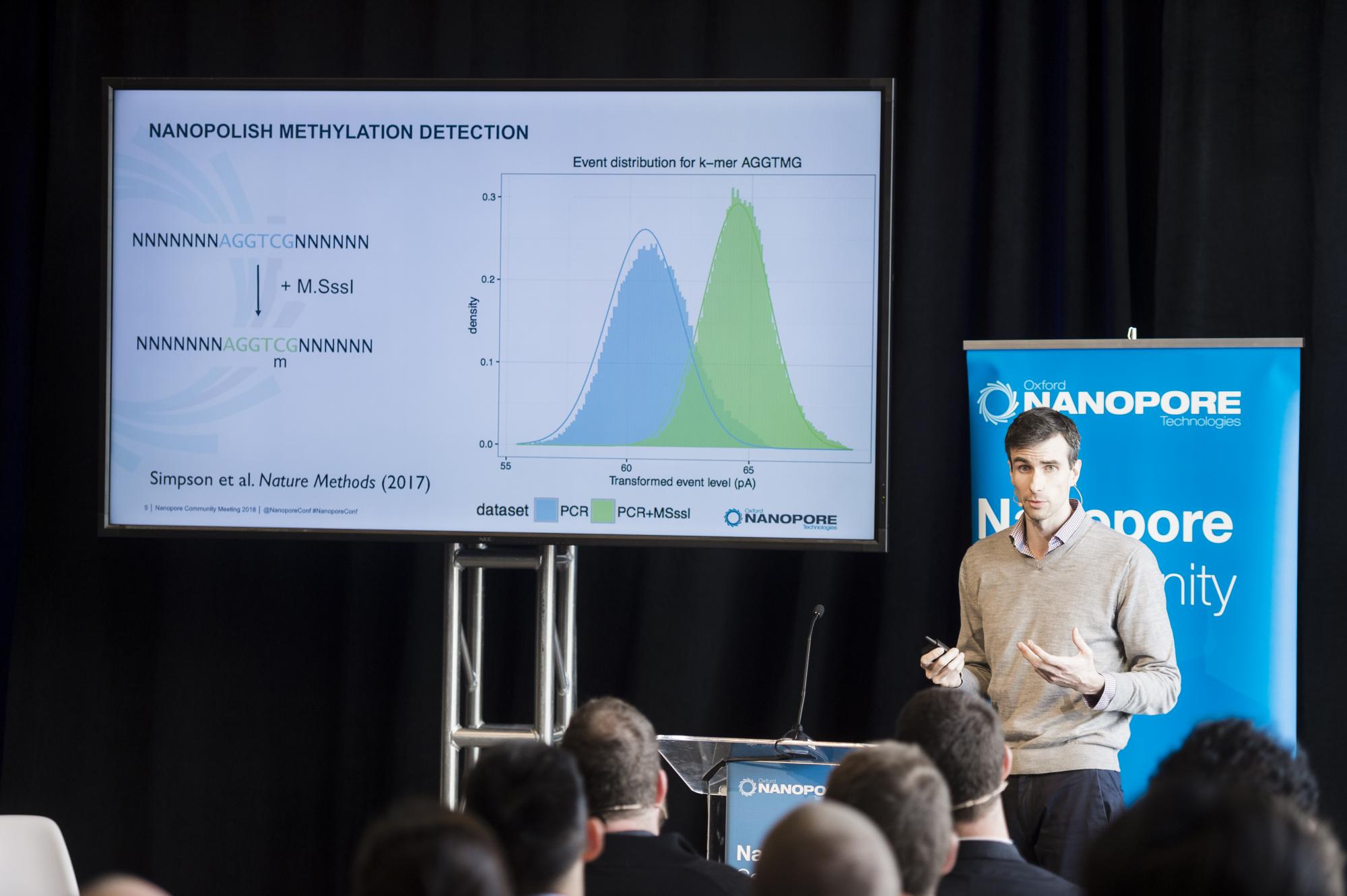
Jared then moved on to consensus calling. The tool uses a hidden Markov model (HMM) to examine the raw data for possible improvements to the consensus sequence. In collaboration with Nick Loman and Josh Quick, Jared has used nanopolish to calculate consensus sequences from a mock microbial community, containing 8 bacterial and 2 yeast genomes. When used on reads obtained using PCR-based library preparation, nanopolish (after miniasm and 2 rounds of Racon polishing) significantly improved assembly accuracy. For native DNA, Jared noted that E. coli and Salmonella concensus calling accuracy didn’t improve as drastically as for other species. This is due to the high level of methylation in these two organisms. In light of this, Jared built methylation aware-polishing into nanopolish, which significantly improves the consensus calling of methylated DNA. Closing his presentation, Jared shared data generated just this week, on the use of nanopolish with data obtained by combining the R9 and R10 pores, which revealed over 99.9% consensus accuracy.
Ian Holmes
In the second presentation from the Raw signal analysis breakout, Ian Holmes from the University of California, Berkeley provided an informative overview of how dynamic programming techniques are key to many nanopore basecalling and analysis. He started by showing a tweet from the London Calling conference which implied that hidden Markov models are old technology, however Ian countered that they are intimately linked with recurrent neural networks (RNN) and there is still an active role for them in basecalling. Ian described how his team are using automata theory (understanding and simplifying how machines compute) to bring together different algorithms such to deliver enhanced results.
He elaborated on how his team at UC Berkeley are developing Machine Boss, a tool for combining modular state machines (i.e. a way of building up a more complex machine from relatively simple components). One challenge of this combining modular state machines though is the huge increase in the number of states. Ian revealed that by keeping only the most likely paths, they are able to drastically reduce the size of the automata. He showed data revealing that Machine Boss, when used to combine the output of protein sequence and base calling, delivered a 12 bit improvement in signal. He also described another new tool from his group, called PoreOver. This new basecaller incorporates a nanopore-basecalling RNN and can also accept the output of other similar RNNs. The tool has been trained on large volumes of publically available data. Ian shared data revealing that the use of PoreOver on 1D2 reads resulted in a 5% increase in read accuracy. Both Machine Boss and PoreOver are available on GitHub.
Marcus Stoiber
In a last-minute change to the agenda, Marcus Stoiber gallantly stepped into the breach to discuss the latest developments in modified base calling. He opened his presentation by describing Tombo and a series of improvements that have recently been implemented. The Tombo analysis platform utilises raw signal analysis and basecalled reads to allow detection of epigenetic modifications from raw nanopore sequencing data. Three different ways of detecting modified bases are available, providing users with flexibility with regard to application and experimental design.
Marcus explained that recent work on the generation of a number of motif-specific models (in human and E. coli) has dramatically improved the power of the approach and delivers more accurate calls. He also highlighted the availability of online tutorials that allow researchers to train the tool to detect base modifications in their organism of interest – providing it has a specific sequence motif/pattern. A further improvement to the tool is the introduction of ‘level sample comparison’ which allows improved modification detection in specific contexts, such as highly modified direct RNA.
In the next section of his talk, Marcus touched on the ‘flip-flop’ algorithm that Clive Brown revealed in his plenary presentation on day 1 of the meeting. The flip-flop algorithm simplifies base calling by outputting single bases as opposed to more complex k-mer labels. This provides a better architecture for calling base modifications, allowing the output to scale gently with the addition of different bases. Training the model is much easier and requires less data. He also provided data showing how flip-flow successfully called 5hmC with no loss in accuracy to the underling base.

Breakout: RNA - Expression
Jonathan Goke - Beyond Gene Expression: Long Read RNA-Sequencing of the Cancer Transcriptome
Johnathan Goke, a PI from the Genome Institute of Singapore, gave a presentation outlining the ways he and his team had systematically evaluated the use of the; PCR cDNA, direct cDNA and direct RNA sequencing kits provided by Oxford Nanopore Technologies, using cancer cell lines. After giving an overview of gene expression mechanisms and applications, such as studying pluripotency, Jonathan explained how transcriptomics is rarely a case of one gene, one product. Alternative splicing and the generation of multiple isoforms from a single gene significantly increases the complexity of this system. Giving an example of why this was important, the ERBB2 gene, a known oncogene, has two promotor regions. A significantly higher rate of mortality if seen if the second promotor region is activated. This lead Johnathan to say gene expression alone is an insufficient simplification.
Moving on Jonathan said how this talk was mainly going to be an assessment of the current technology available and the three kits provided by Oxford Nanopore Technologies, namely the PCR cDNA, direct cDNA and direct RNA kits. Furthermore, some comparisons with short read technologies would be shown. Jonathan described how currently short read technologies used to generate RNA based sequencing data result in systematic biases and that long read solutions, particularly those which are amplification free have the potential to overcome the major limitations of short read sequencing.
Introducing the concept of nanopore RNA sequencing, Jonathan outlined the fact that Nanopore provides methods to produce full length RNA sequences via both direct sequencing of the RNA or via a cDNA intermediate with or without the use of PCR. As part of the Singapore nanopore-expression consortium, Jonathan’s aim was to generate nanopore data from six cell lines, establish QC and bioinformatic workflows to analyse the resultant data and study patient to patient variation in order to detect novel RNA biomarkers.
Outlining the advantages of each kit, including new developer versions, Jonathan produced a table stating that the PCR cDNA kit had low input (1ng) and high throughput but required PCR and did not detect modifications. Furthermore, he had access to early chemistry changes provided by Oxford Nanopore which produces stranded data producing sequences in the 5’ – 3’ orientation of the original RNA molecule. Moving on, the direct cDNA approach has a higher input requirement (100 ng) with medium throughput but does not require PCR. The direct RNA was then described as having the highest input requirement (250ng) than the other kits but had the distinct advantage of maintaining information about modifications in the signal data. Jonnathan said that the average read length for the kits were all over 1000 bp and direct cDNA produced the longest reads. There was high technical reproducibility between kits and samples and any subtle differences could be removed with a linear model-based approach.
Next Jonathan presented data describing a direct head-to-head of these nanopore kits with short read technology-based approaches. He said that a depth of 1 million reads enables the detection of 30, 000 transcripts and there were very high correlations between the short read technology and all the long read approaches when gene level analyses were performed. Jonathan then said this provided evidence to suggest that these kits could be “backward compatible” with previous experiments using short read data alone.
Jonathan went on to show that the 3’ bias he detected in his short-read data was significantly reduced when using the nanopore long read sequencing methods. Stating “as someone who has worked a lot on RNA sequencing this [even coverage] was very pleasing.”
In addition, when examining the number of splice sites covered by single reads, very few of the short reads data spanned one or more splice sites while this was significantly increased in all bins for the long-read data. Furthermore, unlike the short-read data, most of the sequences generated by the nanopore methods uniquely mapped to single transcripts references. As a result, although the gene level correlations between the two technology platforms were remarkably good, the transcript level correlations were significantly reduced.
Jonathan finished his talk by stating that all the data from the SG-NEx will be made public and summarised his finding by saying nanopore RNA-seq has high consistency across replicates protocols and platforms and that although biases are present these are significantly reduced compared with short read data.
Anthony Bayega - Transcriptome landscape of the developing olive fruit fly embryo delineated by Oxford Nanopore long-read RNA-Seq
The olive fly is a non-model organism which is under-studied, however is one of the most important pests for cultivated and wild olive fruits costing over 200 million dollars in crop loss. A diploid organism with a genome of 450 Mb and 6 chromosomes, there are a number of developmental mechanisms that are characterised by dramatic transcriptional changes over short time periods. Anthony explained how sex determination occurs within the first 6 hours of embryo development and is mediated through alternative splicing events, however the male-determining factor remains elusive.
Using nanopore long read technology, Anthony stated that his aims were to identify complete transcripts and perform de-novo transcriptome assembly and compare these to current gene models predicted by NCBI. Furthermore, he said his final aim was to attempt to delineate temporal transcript kinetics that occur in the first two hours of development.
Using a time series experimental design, total RNA from olive fly embryos at 1 – 6 hours post oviposition was extracted and reverse transcribed using a strand switching approach. Using LSK-108 ligation sequencing kit a total of over 31 million cDNA reads were generated for the entire experiment. Comparing sequences from this approach with cDNA sequences generated from short read technology, forty-fold fewer ONT reads were required to detect the same number of genes, while 7.9 fold fewer bases were required. The two technologies agreed in terms of gene counts with a Spearman’s rho value of 0.739 and showed that the majority of transcripts spanned the 5’ – 3’ end of the RNA references used.
Anthony went on to show how the long reads generated in this experiment could be used to correct mis-annotated genes and that direct absolute normalisation of this RNA data, using ERCC spike-ins, out performs relative normalisation techniques commonly used in these types of experiment. Furthermore, when the mRNA concentration was calculated from sequence data over the first two hours, the theoretical concentration halved. This was validated by qPCR suggesting that patterns in the sequencing data reflected the actual abundance of mRNA transcripts as defined by qPCR.
Next Anthony showed that transcriptional expression profiles for each time point over the first 6 hours of development could be compared in a pairwise fashion, and that those closer in time showed a higher correlation that those of distant time points, identifying clear transitions in developmental processes taking place. To explore temporal patterns in higher detail, transcripts were clustered using absolute expression profiles over the time course. Specific clusters containing genes used in early development could be seen decreasing in abundance over the course of the experiment, while those involved in late developmental processes could be seen to be up regulated at later time points.
Moving on to the last section of his talk, Anthony described how long read nanopore sequencing was used to improve the annotation of the Doublesex gene over that already generated by short read sequencing. To highlight why understanding the mechanisms of sex determination in this organism is important, Antony showed how a CRISPR-Cas9 targeting the doublesex gene in caged mosquitoes showed that complete population suppression could be achieved, suggesting this is the perfect target for population suppression in the olive fly. As a final highlight slide, Antony mentioned that he has been an early developer of the up-and-coming PCS-109 PCR cDNA sequencing kit showing that this kit provided a simpler workflow and has doubled the throughput of their sequencing runs.
Paolo Poggio - PCSK9 knockout mice are protected from valvular calcification
Paolo Poggio, from the Monzino Cardiology Center in Italy, gave a talk examining the effect of the gene PCSK9 on aortic valvular calcification using a knock out mouse model. Paolo started by giving some background into why they chose Oxford Nanopore Technologies a s a sequencing solution. Having never performed any sequencing before, but having a grant deadline looming, a quick and simple method to go form question to answer was required. After a training session at Oxford Nanopore Technologies, Paolo and his team had sequencing data within two weeks of concept. Moving on he began to describe the disease under study in his presentation.
Aortic valvular calcification manifests as accumulated calcium deposits on the aortic valves of the heart. This results in reduced blood flow and is associated with significant cardiovascular morbidity and mortality. Approximately 3 % of the population are affected by the disease and there are currently no pharmacological interventions with invasive surgery being the only option. In addition, if surgery is not undertaken the life expectancy does not exceed 5 years. Although there is no pharmacological intervention available, there is evidence to suggest that proprotein convertase subtisilin kexin type 9 (PCSK9) inhibitors may reduce disease onset and progression through regulation of calcium deposition.
In terms of experimental design, Paolo described how a knock out mouse model was used to measure the total aortic calcium content and compared the values to the wild type control. There was a significant reduction in total calcium content in the aortic valves (p < 0.001) and specific cells, known as valve interstitial cells, has significantly lower calcification rates (p < 0.001). In addition, calcification was induced chemically in this system to show that these levels could return to just below control levels.
A differential gene expression study was designed in order to determine which genes were up- and down-regulated in the knock out mice when compared with the wild type controls. Using Oxford Nanopore's PCR cDNA kit, more than 3 Gb per run was generated, resulting in the detection of > 200 differentially expressed genes with over a log2 fold change in abundance and corrected p values of < 0.05. Triplicate samples were multiplexed across single flowcells allowing for large changes in dominant transcripts to be detected.
When performing a functional analysis of these differentially expressed genes, upregulated genes were associated with the p38 cascade and cytoskeletal modification, while downregulated genes regulated apotopic pathways, cell adhesion and oxidative stress responses, a lot of these being related to PCSK9 pathways.
Showing that a link between PCSK9 and aortic valvular calcification in a mouse models using both direct measures of calcification and Oxford Nanopore long read cDNA sequencing, lead Paolo onto obtaining a grant from the European Research Area Network on Cardiovascular Diseases in order to study this process in humans.
Using human cells isolated from aortic valve replacements, Paolo showed that PCSK9 was significantly up regulated in aortic valvular calcification samples when compared with controls. As PCSK9 expression is related to oxidative stress, cells were exposed to an oxidative stress via the addition of H2O2 and calcification rates increased.
Paolo finished by saying that based on the results of these experiments, they now have the opportunity to test if pharmacological inhibition of PCSK9 could reduce or even halt the progression of this wide spread and debilitating disease.

Breakout: Structural variation in cancer
William Jeck: Nanopore Sequencing and Rapid Fusion Testing - a "Killer App" in Molecular Pathology.
William Jeck, of the Massachusetts General Hospital Department of Pathology, described his team’s use of nanopore sequencing as a ”killer app” for molecular pathology: the rapid detection of oncogenic gene fusions in clinical samples.
William demonstrated the challenges of quickly diagnosing and treating acute leukemia and sarcoma by outlining a clinical case for each. The first was of a man who was diagnosed with acute promyelocytic leukemia following cytogenic testing, 48 hours after referral to the emergency room; by this time, a treatment which specifically targets the PML-RARA translocation responsible for acute promyelocytic leukemia had been pre-emptively started. William asked: can we identify the presence or absence of the PML-RARA translocation faster than this? This would enable more rapid diagnosis-to-treatment, and prevent time wasted on an incorrect treatment. The second case was that of a man whose diagnosis of atypical Ewing sarcoma took 16 days and several methods of testing: immunohistochemistry analysis (identified unclassified round cell sarcoma, but did not allow a more precise diagnosis), targeted testing for CIC translocation (returned negative after one week) and EWSR break-apart FISH analysis (positive for rearrangement). William asked: can we detect soft tissue-related fusions in the same timeframe as immunohistochemistry testing?
William highlighted how “Nanopore sequencing fits perfectly with the needs of clinical fusion detection”: the MinION fits easily into hospital laboratories which are stretched for space, where real-time sequencing has potential to provide rapid diagnosis at a cost within the range for a clinical test. He noted that the long-read output “covers fusion breakpoints… and then some”, with throughput generating more than enough coverage for fusion calling.
The team developed a pipeline to test for gene fusions in clinical samples using Anchored Multiplex PCR (AMP) and nanopore sequencing. RNA was extracted, reverse transcribed and ligated to adapters; this was then amplified via AMP, using a nested PCR with both adapter-specific and gene-specific primers to enrich for the regions of interest. The enriched samples were then amplified with Oxford Nanopore’s barcoded primers and sequenced in multiplex on the MinION device. Reads were aligned via BWA-MEM to a synthetic transcript reference library constructed from existing transcript annotations (William noted here that he would like to use minimap2 in future); the resulting dataset allowed for both the identification and quantification of fusions. The pipeline was first used to sequence an erythroleukemia cell line, in which it successfully identified a BCR-ABL1 fusion, with fusion reads being generated within seconds of starting sequencing – William described how he “stopped the run within ten minutes because I knew we had a success” and was then able to reuse the Flow Cell. He demonstrated how the long nanopore reads resolved long-range exon structure across the fusion.
The workflow was then put to the test on clinical samples: a PML-RARA fusion was identified in a leukemia patient sample, whilst a HAS2-PLAG1 fusion was identified in an FFPE lipoblastoma patient specimen. A blinded test of the fusion-calling workflow was then performed, using 11 fresh hematologic specimens and 5 FFPE sarcoma specimens. From these, 12 true positives and 3 true negatives were identified. Following four false-negative calls, with two due to the low fraction of tumor DNA present in a qPCR validation sample used (representing less than would generally be seen in a clinical sample), the team then investigated the sensitivity of the test. Samples containing a varying fraction of tumor DNA were sequenced; fusion genes were identified in samples containing down to a fraction of 5% tumour DNA.
William concluded that nanopore sequencing of AMP-prepared libraries successfully identifies oncogenic gene fusions, rapidly and with high sensitivity, in both fresh and FFPE samples; he highlighted that this demonstrates the technology’s potential for use as a rapid clinical diagnostic test, given suitable validation for this purpose. Lastly, William revealed that he had recently sequenced, to his knowledge, the first clinical sample to be run on a Flongle Flow Cell; he was able to identify an ELBR-FLI1 gene fusion, showing its potential as a single-sample, affordable diagnostic tool.
James Blachly: Real-time leukemia diagnostics with nanopore
Note: James provided disclaimers regarding his consultancy work for pharmaceutical companies, discussion of “off-label” use of medications and participation as a sub-Investigator in clinical trials of the agents discussed; please refer to his presentation for more information.
James Blachly, MD, of The Ohio State University, presented his team’s work demonstrating how nanopore sequencing could enable real-time, same-day molecular diagnostics for acute myeloid leukemia.

James described how acute myeloid leukemia (AML) is the most common form of leukemia in adults, and typically requires treatment via intensive, month-long in-patient chemotherapy; deaths sometimes occur during induction chemotherapy. AML represents “one of the few potential oncological emergencies”: rapid diagnosis and commencement of treatment is of very high importance. He notes that the genetic drivers of the disease have been “well understood for decades” and provide important prognostic information, but have only recently become targetable; whilst a vast amount of data was available for point mutations and cytogenic abnormalities, no new drugs were introduced for forty years (between 1973 and 2010, with the introduction of Gemtuzumab). This changed in 2017-8, as several new targeted therapies were made available (James noted that another new drug was released the day before this talk), designed to target disease associated with specific mutations; James focused on drugs targeting recurrent mutations in FLT3, IDH1 and IDH2.
James explained how new AML patients are frequently referred to academic medical centres such as Ohio State, which specialises in leukemia and has rapidly adopted the newly available targeted drugs midostaurin, enasidenib and ivosidenib; however, outside of such institutions, uptake of these new drugs has been limited. This is due to the need for molecular diagnostics to elucidate the mutation present, in order to identify the suitable targeted treatment. For this, many institutions send off samples for testing with broad next-generation sequencing panels, but this can take over a week – a timeframe which, without treatment, “can be life-or-death for patients with AML.” He compared the current diagnosis-to-treatment timeline, in which NGS results are received around day 9-12, with the timeline he and his team envision: diagnosis, sequencing results and ordering of targeted treatment on day 1. Their goal is to use automated library prep via VolTRAX, followed by rapid sequencing on MinION or Flongle Flow Cells, to test for AML using peripheral blood samples.
The team used the PCR Barcoding Kit (SQK-PBK004) to prepare AML samples for nanopore sequencing in multiplex on the MinION device; James noted that if this was developed for use as a clinical test, they would prefer to avoid multiplexing in favour of single-sample testing via Flongle. They then used hotspotter, their cloud infrastructure for variant analysis and reporting, for subsequent identification of mutation hotspots in the data. James showed FLT3, IDH1 and IDH2 mutations successfully identified in data from 48 targetable lesion samples via nanopore sequencing; these same mutations were identified via two short-read technologies, demonstrating “perfect concordance” between the datasets, with 100% sensitivity and specificity.
Lastly, James demonstrated that nanopore sequencing enabled the detection of internal tandem duplications in FLT-3 inhibitors, which are seen in 25-30% of AML patients. These repeats can be 15-300 bp long, and occur within a three-exon area; whilst long nanopore reads were able to span these duplications for effective detection, they were detected poorly by short-read sequencing.
James concluded by stressing the importance of rapid molecular diagnosis in identifying suitable targeted AML treatments for patients as soon as possible; nanopore sequencing, he highlighted, is “ideally placed for this”. Their workflow could potentially reduce diagnosis-to-treatment time by a week or more, and enable molecular diagnostics to be performed at community oncology practices: “we can democratise the treatment of leukemia.” The team’s cloud infrastructure will be available in 2019 and their hotspotter software in January.
Therese Woodring - In Cold Blood: whole genome sequencing in a fatal septic transfusion reaction
Therese Woodring, of the University of Illinois College of Medicine, Peoria, described the use of Oxford Nanopore’s MinION device in investigating the organism behind a fatal septic transfusion reaction (STR). She outlined the case of a woman who received a blood transfusion: after five minutes, the nurse could see that the patient’s breathing had changed and heart rate had increased, so stopped the transfusion. A transfusion reaction was suspected ~2 hours later, and Gram-negative rods were identified in the patient’s blood ~3 hours after this. After another ~5 hours, the patient was transferred to intensive care, and an hour and a half later was pronounced dead. Therese noted that fatalities from STRs are remarkably rare – in the single figures, despite the ~5 million transfusions performed each year, a credit to the work of blood banks, and also due to the inhospitable environment of blood packs. She described how, for example, the packed red blood cells used in transfusions are stored at 4C and accumulate waste products and reactive oxygen species – “not exactly a gentle culture media”. To investigate the kind of organism that could exploit this extreme enrivonment, Therese and her team performed whole-genome amplification of isolate DNA samples. Libraries were prepared using the Rapid Sequencing Kit (SQK-RAD004) and sequenced on the MinION device. Analysis was carried out on Therese’s laptop: assembly via Canu produced a 7.34 Mbn chromosome; 16S identification via three database searches indicated Pseudomonas, with two specifying P. poae, a ubiquitous soil bacterium often found in cold environments, including the Himalayas – not a human pathogen. Multilocus 16S analysis also placed the isolate in the P. fluorescens subgroup. Several cold tolerance genes were identified in the sequenced isolate; cold tolerance testing revealed growth at 4C and 25C, whilst the bacterium was killed at body temperature. This indicated that its pathogenicity in this case was an ecological problem, unlikely to occur outside of this environment. The team also identified four siderophore receptor genes, important in the acquisition of iron, which produced fluorescence under UV light. Therese concluded that whole genome sequencing of the isolate on the MinION helped to clarify the identification of the bacterium and characterise its behavioural repertoire – “a non-pathogen rendered lethal in cold blood” - whilst also improving understanding of the ecological context of STRs. The team hope their work will help inform future STR isolate investigations and encourage a “DIY” approach to the use of sequencing in medical education.

Paul Gordon: On the motion of planets, and nanopore signal consensus.
Paul Gordon, MCS, PhD, of the Centre for Health Genomics and Informatics, University of Calgary, described the use of Dynamic Time Warping Barycenter Averaging (DBA), an algorithm used in aligning two sequences to each other, on direct RNA squiggles generated in nanopore sequencing. Using the squiggles generated by yeast enolase, Oxford Nanopore’s RNA sequencing spike-in, he demonstrated several methods of generating alignments: symmetric vs asymmetric, batch vs incremental. He notes that whilst the symmetric batch method is currently considered computationally infeasible, an asymmetric incremental approach has become possible just recently. Paul evaluated the use of three implementations of DBA, which each produced different results – he highlighted that one algorithm, SSG, produced results which were longer than the signal due to spurious events; and explained that a custom voting system can be used to remove these. This approach can be used to align nanopore squiggles to each other in the absence of a model, enabling identification of modified bases without the need to generate a training dataset.
Sara Goodwin: Exploring the architecture of organoid genomes with PromethION technology.
Sara Goodwin, of Cold Spring Harbor Laboratory, described how long-read nanopore sequencing can be used to resolve structural changes that occur during the generation of organoids. Organoids are three-dimensional cell cultures, which can be grown from cells taken from tumour biopsies; where biopsies can yield little DNA for long-read sequencing applications, organoids can generate sufficient material. A relatively new technology, they are used in a range of applications including the study of disease genetics & pathology and drug screening, and provide “an important tool in cancer biology.” Sara described how DNA from organoids derived from breast tumour, normal breast tissue and the breast cancer cell line SKBR3 were sequenced via nanopore sequencing, a short-read technology and a long-read technology. Sara displayed structural variant calling for the patient tumour organoid DNA: results were highly concordant between the two long-read technologies, whilst the proportion of data called as structural variants in only the short-reads dataset were shown via PCR validation to mostly comprise false-positives. She went on to show a 62 bp repeat expansion in a BRCA1 intron, which was successfully detected via long reads, but not via short-read sequencing; this was also the case for a ZMYM3 50 bp deletion and an NF1 100 bp insertion. Sara then demonstrated how her team were using nanopore sequencing to investigate methylation: “huge hypermethylation” was seen in the normal sample, whilst correlation of methylation patterns between SKBR3 and the tumour organoid suggested the presence of cancer-specific methylation.
Piroon Jenjaroenpun: Uncovering RNA modifications from native RNA sequences using Eligos
Piroon Jenjaroenpun, of the University of Arkansas for Medical Sciences, described the development of ELIGOS: a software for the identification of RNA modifications in nanopore direct RNA sequencing data. He described how there are over 100 chemical modifications in RNA; whilst there are several methods of profiling these modifications, each enriches only one specific type of modification at a time. Oxford Nanopore’s direct RNA sequencing data, however, preserves all types of RNA modification via direct sequencing of native RNA strands; these can be elucidated bioinformatically from “glitches” in the signal in sequencing. Piroon and his team developed ELIGOs, Epitranscriptional Landscape Inferring from Glitches of ONT Signals, a simple workflow for capturing multiple RNA modification types. RNA samples are sequenced using both direct RNA sequencing, in which native RNA modifications are intact, and direct cDNA sequencing, in which modifications are not present. Specific base sites which are frequently called in direct RNA as insertions, deletions and substitutions, but not in cDNA, are identified via a Fisher’s exact test, to determine likely modified base sites in the native RNA sample. The team used ELIGOs to capture modifications in yeast rRNA, identifying 95% of the modified bases previously elucidated by Yang et al. (Yang (2016) PLoS One. 2016; 11(12): e0168873) using RP-HPLC and a mung bean nuclease-based assay. Piroon also demonstrated the use of ELIGOS in the identification of differential proportions of m6a modifications in the junB proto-oncogene in samples of RNA from human embryonic kidney cells, CD8-positive cells and lung adenocarcinoma epithelial cells.
Amy Klink: Nanopore (MinION) sequencing to detect and sequence full genomes of emerging RNA viruses in avian and marine mammal species
Amy Klink, of the University of Alaska Anchorage, described how she and her team used nanopore cDNA sequencing to rapidly genotype emerging avian and marine mammal viruses throughout regions of Alaska. Amy noted that RNA viruses are of particular interest: they are able to rapidly mutate, and can jump between species and between host cells. She described how viral outbreaks of two Paramyxoviruses - Newcastle disease virus (NDV) & phocine distemper virus (PDV) - and influenza had resulted in the deaths of over 1,500 harbour and grey seals off the Anchorage coast. Amy and her team first adapted an MS-RTPCR protocol for avian and mammalian influenza to generate sequencing libraries from primary preserved influenza samples. The paramyxoviruses NDV and PDV were amplified via a Zibra project tiling RT-PCR method. The resulting partial- and full-genome amplicons were prepared for sequencing using the 1D^2 (SQK-LSK308) and 1D (SQK-LSK108) kits and sequenced on the MinION. Reads were basecalled via Albacore, trimmed with Porechop/ Filtlong and mapped via Minimap2. Amy displayed how the NDV 1D^2 library provided initial proof of concept, producing a pairwise identity with the LaSota strain of 74.4%. As the team generated more sequencing data for the genomes, they were able to map where the viruses were evolving from: analysis of the segmented genomes of influenza isolates demonstrated reassortment of segments from very different areas, enabling the mapping of potential outbreak hotspots.
Cameron Soulette: Nanopore Sequencing reveals isoform-specific changes associated with U2AF1 S34F.
Cameron Soulette, of the University of California Santa Cruz, discussed his lab’s use of nanopore cDNA sequencing in the identification of cancer-specific mutations in the splicing factor U2AF1. He explained how U2AF1, which plays an essential role in defining the 3’ end of introns, is recurrently mutated in several cancer types, including in 3% of lung adenocarcinomas. He focused on the most common mutation, S34F, which produces significant alternative splicing events in lung adenocarcinoma. This results in the formation of aberrant mRNAS; however, the functional impact of these splicing alterations is poorly understood - Cameron questioned whether this variant could be selected for in lung adenocarcinoma due to it conferring an advantage to tumour progression or maintenance. He noted that characterisation of isoforms with short reads is difficult; the team used nanopore sequencing to generate long-read sequencing data for these isoforms. cDNA libraries were generated in triplicate for samples of two wildtype cell lines and two mutated cell lines; these were then sequenced on the MinION device. Data was also generated using a short-read sequencing technology. Full-length isoform analysis of RNA (FLAIR) was performed to characterise the isoforms present, revealing alternative splicing proportions for the wildtype vs S34F samples. Of the S34F-associated isoform changes identified, 30 were identified by FLAIR with long-read nanopore sequencing only, 3 by the short-read assembly only, and 5 by both methods. Of the significantly dysregulated isoforms identified, two thirds were not seen via short-read sequencing and were missing from the reference annotation, suggesting novel isoforms. Cameron then displayed data for UPP1 isoforms, associated with an unfavourable outcome in lung cancer: here, complicated, long-range isoforms were picked up by FLAIR analysis with long nanopore reads but not by short-read assembly. Cameron concluded that the use of long reads here enabled the identification of cancer-related genes for further functional analyses.
Spotlight session
Thursday afternoon saw the second Spotlight Session of the conference, where early career scientists are given just two minutes on stage to pitch how sequencing is changing their field of research before the audience vote on who gets to take the spotlight for a main plenary talk. The other two speakers still get to present though, just in the following Mini Theatre session.
Initially planned for just the one session, the Spotlight Session was featured on both days instead due to a huge number of excellent submissions.
First on stage to pitch their talk was Roxanne Zascavage, who opened by stating that she didn’t need two minutes to convince the audience that her talk would be the best. Asking the audience the identity of a photo on her slides (the Golden State Killer), Roxanne outlined that her talk would provide mass disaster, serial killers, and why your relatives are making it more difficult for you to get away with murder, before declaring the end of her pitch – to a loud round of applause!
Following Roxanne to the stage was Nicola Hall, who presented a hypothetical patient, Alfie, to demonstrate the problem that psychiatry has – that of diagnosis. In this example, Alfie had schizophrenia, and started to have unusual experiences, hearing voices that weren’t there, before onset of paranoia and psychosis, an episode of which lead to Alfie ending up in emergency care.
After admission to hospital, Alfie would be given an antipsychotic drug, but the side effects of this include sluggishness and weight gain, and while the drugs would help Alfie deals with his symptoms, they would do nothing to target the root cause. In particular, the diagnosis of schizophrenia specifically would take considerable time, as symptoms for schizophrenia are shared amongst many other psychiatric disorders. While Alfie’s symptoms are treated, he would continue to experience problems with cognitive and social functions. Better treatment is clearly required, Nicola explained, to understand the underlying biology and so treat patients effectively and suitably. Nicola’s talk would feature investigations into the expression and splicing of a psychiatric risk gene, conclusions from which could lead the field forward to new treatments for patients like Alfie.
The final pitch came from Thidathip Wongsurawat, who posed the question: what should we do where vaccines and antiviral drugs are available, but RNA viruses continue to infect people and spread across the world no matter what? Tip explained that her group aimed to accelerate viral research by developing something applicable to any income country – a good, fast and cheap way to do RNA viral sequencing. The strategy, Tip said, was very simple – using direct RNA sequencing to capture several layers information. It would allow sequencing of multiple viruses from different families with no barcoding, on a single flow cell with no primers and no reverse transcription required. Tip finished by saying that her talk would feature details of strategies and lessons learnt along the way, which would hopefully be informative to the Community.
After a tense vote, Roxanne won with 52% of the vote, with Nicola and Tip finishing on 24% each.

Returning to the stage, Roxanne introduced herself as a forensic DNA investigator, whose work revolved around DNA sequencing technologies for forensic applications. Roxanne’s opening slide featured a quote from “the most famous forensic investigator” – Sherlock Holmes. The quote centred around the identification of blood from a dried sample, but, Roxanne noted, the same quote about changing the game of criminal investigation could be applied to DNA sequencing.
Roxanne went on to discuss the broad range of applications within forensics to which DNA sequencing could be applied – including but not limited to criminal investigation, missing/unidentified persons, paternity tests, immigration disputes, bioterrorism and familial searching.
Moving to the history of DNA testing in forensics, Roxanne described the first utilisation of these techniques in the UK in the mid 1980s, to exonerate an innocent man and identify and convict another, before broader expansion to the US in 1987 and the DNA Identification Act in 1994 that allowed suspects to have samples collected and information logged in a database.
Putting this into context and taking a step back from DNA specifically to all evidence, Roxanne showed a picture of the 9/11 disaster, where a team deployed immediately following the event processed 21,000 remains in an attempt to identify the several thousand missing persons. In terms of evidence, Roxanne posed the questions: what happens to evidence when it is burnt? Left out in the sun? Or discovered many years later? Most evidence, Roxanne explained, is degraded and rendered useless – but mitochondrial DNA perseveres making it an ideal candidate for analysis of evidence. Of the 9/11 individuals that have been identified, the last three were named in 2015, 2017 and the final just two months ago, demonstrating that as technology advances and nucleic acids can be analysed even at very low abundance, more and more cases can be resolved. Connecting that mitochondrial DNA to those individuals has been critical to the family and loved ones of the missing people.
Changing tone from disaster to serial murders, Roxanne reiterated that as long as we have remains we can work towards identification. However, databases mostly currently include only data from those that have been convicted, although these rules vary by state - some include those that have been arrested or permit familial searching.
Roxanne then asked: what do you think of when you think of a serial killer? A white male? Databases in general are heavily biased towards minorities, and this inhibits isolation of suspects. The ideal would be, Roxanne states, to take a sample from a crime scene, run an analysis from start to finish, and obtain all the information you need – but often this is limited by references to identify against. Voluntary databases, though, represent the beginning of a solution to this – as Parabon has been used to solve 20 cases so far, including cold cases. Public databases have no regulation, and this could lead to concerns over safety.
In two years, Roxanne concluded, 90% of Caucasians will be able to be identified by cousins, aunts, uncles, sisters, brothers using these websites and publicly accessible data.
Panel plenary
Martin Smith: High-throughput targeted nanopore sequencing of single cells
Martin began by introducing the fact that him and his team have been doing nanopore sequencing for a while, encompassing applications in cancer genomics and epigenomics, transcriptomics and RNA modification detection, as well as a small amount in the production sequencing space.
For this talk though, Martin focussed in on single cell sequencing, which can be likened to population genomics for tissues rather than across a population of organisms. Lots of platforms, Martin explained, can do single cell analysis, but not many (if any) can do both UMIs alongside simultaneous generation of full-length transcripts. The single cell platforms Martin’s team use are droplet-based, as these have high throughput in comparison to plate-base or flow cytometry techniques. In particular, Martin and team use the 10X Chromium device to isolate individual cells into reaction droplets. The basic workflow is to homogenise the tissue sample, capture cells into droplets and perform reverse transcription on the beads, before amplification with PCR to generate a cDNA library.
Traditionally the next step would be to chop the cDNA up into fragments ready for short read sequencing in order to generate 3’ expression profiles, which can then be used for declustering.
The focus of doing this kind of analysis though, Martin explained, was to examine how the immune system of a patient keeps cancer in check, or what is it that causes cancers to evade the immune system? In order to understand oncoimmunology, Martin’s team have described the “holy trimmunity”; namely sequencing of tissue from the tumour, lymph node, and blood of a single cancer patient.
Each lymphocyte has a unique antigen receptor sequence, and these undergo massive somatic recombination, making it extremely unlikely that two cells have the exact same combination of exons. The primary limitation of short reads, Martin outlined, is that they only return one end of these molecules, only capturing the 5’ or 3’ (beginning or end) of the transcript.
The solution to this limitation though is perhaps nanopore sequencing, although the per read error rate must also be taken into account when pulling out short barcodes of approximately 16 nucelotides in length for accurate demultiplexing. Martin and the team at the Garvan Institute have developed a process called RAGE-seq for this purpose, in which a normal cDNA library is prepared with the droplet method and submitted for gene expression profiling. In parallel, though, capture probes have been designed to pull out sequences for T and B cell receptors, with which the short reads are combined to identify the barcodes accurately. The resulting contigs are assembled de novo, to find out which VDJ sequence is present.
This process was benchmarked with three well-characterised cell lines. Clustering plots show that short reads generate the correct proportions of sequences against the proportions of the cell lines put in, so they can be used effectively to pull out the corresponding sequences in the long read data.
Thusfar the process is slightly inefficient, giving 18% recovery of the barcodes, but this can be doubled to 40% with fuzzy matching allowing for a couple of mismatches. Martin explained that as per a previous, raw signal data will be used to demultiplex – but the audience should stay tuned for more progress on that.
The results of the benchmarking on well-characterised cell lines didn’t render a lot of full length RNAs, so some de novo assembly and polishing was required to generate contigs of the expected size of the full mRNA sequence.
These initial results show that 100 reads per molecule is enough to accurately call the clonotype in 50% of cases, and high accuracies of 98% can be generated from just 50 reads. Interestingly, the sequence for IGH and IGL displayed lower accuracy than those of the T cells, but this could be either sequencing artefact or just biology.
Examining the B cell receptor sequences in more detail revealed that the data looked fairly noisy, with lots of variation from the reference. Initially, this was thought to be error, however B cells undergo affinity maturation and somatic hypermutation to finetune antibody sequences so this may not be the case. By way of comparison, Martin performed the same analysis on the sequences of the T cells, which showed almost no mutations in the T cell receptor consensus, so it is likely that the B cells were indeed undergoing somatic hypermutation.
Segueing from here onto patient data, Martin used an example of data from a tumour draining lymph node, containing almost all classes of lymphocyte, where you can identify subpopulations of T cells. Even without looking at the 3’ profiling information, a T cell can be identified as naïve simply based on the number of mutations present in the sequence, and full length contigs from nanopore sequencing allowed the isotype of each antibody to be found.
Martin went on to explain that the applications of the RAGE-seq technique in immunotherapy were both impressive and important for antibodies and antibody therapies, as well as chimeric T cell engineering. Zooming in on T-cell clustering plots allows identification of different subtypes, and this information can be used to track clonal expansion – where all cells that have the same chains begin to spread. Individual lymphocytes clones can be effectively tracked, and Martin presented overlaid data from different tissue types, demonstrating how expression patterns on clonal populations – particularly those with tumour killing properties – can be examined effectively.
This kind of analysis, Martin noted, is also possible on other long read sequencing platforms, but the cost to do this is approximately double that of nanopore sequencing with similar rates of recovery. Concluding, Martin looked to future steps of this research, explaining that the team intend to further develop RAGE-seq ideally to get rid of the use of short read data altogether and generate enough long read data, perhaps with PromethION, to expression profile with long reads. Further than that, removing the capture step and instead looking at the total sequencing output might be another simplifying advance.
Laura Wenzel: Applications of Nanopore sequencing for plant pathogen detection
Laura opened with a discussion of “Why we care about plant health”. With the aim of monitoring and promoting plant health, Laura explained how crop losses through disease cost a huge amount of money and gave the specific example of panama disease. This being a disease in bananas cause by Fusarium oxysporum. This was such a problem in the 1950s the most popular banana cultivar at the time became economically non-viable for production. There are only limited resistant genotypes, but management of the disease is generally restricted to the use of pathogen free planting stock. Plant health across species was important to Laura, from food crops to ascetic angiosperms. In order to monitor and contain plant diseases, pathogen detection in seeds, propagation material and mature plants is needed. Current methods to do this involve using procedures such as indicator plants, ELISAs and qPCR methods. The problem being that many of these approaches work in a targeted fashion, i.e. one must know what to look for. Laura and her team are aiming to use nanopore sequencing to complement or even replace some of these targeted, high skill approaches.
Using an example RNA viral infection in Campanula, a flowing plant, Laura suggested that PCR/antibody-based detection methods could be replaced by sequencing the RNA via cDNA synthesis and taxonomic classification. Campanula can be infected by a number of single stranded RNA viruses, most specifically, Arabis mosaic virus, Cucumber mosaic virus, and Phlox virus S, however there are not defined tests for all potential viral pathogens and thus you can only find what you are looking for. cDNA sequencing using the Nanopore PCS-108 kit and LSK-108 kit reveled that, after taxonomic classification, all three of the above pathogens could be identified, with Arabis mosaic virus sequences dominating the samples. However, Laura pointed out that things got a little strange when HIV virus was also detected and was obviously a false positive. Laura then asked, “why do we get false positives?”. It transpired that a lot of this was due to the sort nature of the query sequence used in the assignment algorithm. Laura suggested that if using something like BLAST the result would be better but as yet she has had little luck. However, using longer hits or splitting the reference sequences up into sections has helped. Moving on Laura showed how she could, as has become a common theme over the course of the conference, detect full viral genomes in single reads. Therefore, again, no assembly was required.
Looking at coverage statistics of some of the sequences from the plant viruses, Laura realized that, one of the plant pathogens of interest, cucumber mosaic virus, was not a poly-A tailed virus and thus was not directly compatible with polyT based reverse transcription priming. Laura stated that random hexamer-based methods could reverse transcribe this virus, allowing correct taxonomic identification.
Laura then moved onto discuss how nanopore sequencing could be used to support current detection methods by generating more complete genomes of plant pathovars so specific genomic detection assays, such as qPCR can be designed to discriminate between closely related but phenotypically different disease-causing organisms. Here a quick example was shown where a bacterial pathogen was sequenced from a lab isolate and was assembled into a single circular contig using 50 K reads and 100 X coverage. This was then compared with 185 contigs produced by 100 x coverage of short reads demonstrating a much more complete genome could be obtained for the same level of coverage.
Charles Chiu: Clinical Sequencing of Pathogens and Human Host Responses in Acutely Infected Patients
Charles began his talk by describing how his lab is a translational laboratory and aims to bring research into the clinic. However, he noted this was commonly known as “the valley of death” as research discoveries into the clinic do not often make it. With a focus on diagnostics, he said that a proposed diagnostic test test needs to be rapid and have a turnaround in a matter of hours rather than weeks to be useful. Furthermore, these need to be able to make diagnoses in critical patients and be able to rapidly detect infection. Charles pointed out that there are often three types of infection that very rarely get a diagnosis, and these are pneumonia meningitis and fever/sepsis. The main problem is that failure to get a timely diagnosis hugely increases mortality rates and financial burden.
Charles then spoke about how they have developed a metagenomic assay that can be used to diagnose neurological conditions using cerebral-spinal fluid as a source of sample. Moving on to the main focus of his talk, Charles proposed the idea that machine learning algorithms could be used as a diagnostic tool and then set out to explain how this could occur. The theory being that, if you fed enough genomic information about all different types of infection, low resolution answers could be rapidly produced in order to quickly inform clinicians at the bed side. Charles said that with the use of sequencing being used routinely in his lab on patent samples, 99 % of the data was being under used and thus was a perfect source of information for model training. Here RNA-seq data was used to train the algorithm. However, Charles pointed out that, generally in a good transcriptome experiment you capture between 60 – 80 % of the transcripts in an organism but he only had about 26 % coverage. He then said he was going to demonstrate why he thought this was enough to provide the low resolution, broad brush answers he required. Suddenly two blurry photographs appeared on the screen and Charles challenged the audience to determine what they were. As they slowly came into focus it became apparent that one of the pictures was of a stylish popular bench top sequencer able to produce ultra-long reads, and the other was a sequencer that…. Wasn’t.
Making his point, Charles said that sequencing data from all different kind of disease could be used as training sets, for example bacterial infections, viral infections, auto immune diseases etc. Using 80 % of the data as a training set and 20 % as a cross validation set a dummy model was used to generate a baseline score of 58 % accuracy. After screening a large number of different model types, it transpired that a radial based function SVM model with feature selection gave 95 % accuracy. Charles said the feature selection part was important as it selected 1000 differential regions of genomic data which discriminated between the different classes of disease. He then said that while this produces great accuracy on a cross validation set, does it work in real cases? Impressively it was able to discriminate between bacterial and viral infections with 91 % accuracy. Next, looking at an unknown case of encephalitis, the model suggested that it was a viral infection with 83 % certainty and it transpired it was a case of Rubella. Next Charles showed how he and his team have created a docker container to run this model locally and used the example of a 2-year-old with a large brain abscess. All clinical cultures were negative, but a successful diagnosis of a bacterial infection was made within 3 hours using the model.
In his closing remarks Charles spoke about a large nanopore sepsis study where he compared a cohort with known bacteremic infection with healthy controls. Showing that good correlations between the ERCC spike in RNA controls he moved on to rapidly discuss some of the findings in the dying moments of his time on stage. Obtaining around 50 % transcriptome coverage per patient, he showed that those with bacterial infections had between 51 % and 73 % probability, according to his model, of having a bacterial infection compared with between 29 and 48 % in the patients in the “other” category. Skipping through his summary slide, one could just make out the statement “Nanopore sequencing coupled with laptop-compatible, rapid analysis pipelines can collect and analyze data in 3-6 hours, a time frame amenable to clinical diagnosis.” before he finished the plenary session and questions began.
Plenary: Todd Michael - Unraveling the mysteries of CBD and THC content with a chromosome resolved Cannabis genome
Closing this year’s Nanopore Community Meeting on a high, Todd Michael, Professor and Director of Informatics at the J. Craig Venter Institute (JCVI) provided an entertaining overview of his team’s research on the cannabis genome. The potential medical benefits of cannabis are of increasing interest to researchers working across a range of disease areas and the demand for non-psychoactive varieties with high concentrations of cannabidiol acid (CBDA) and low concentrations of the highly psychoactive delta-9-tetrahydrocannabinol-acid (THCA) is rapidly growing. The enzymes that produce these compounds, CBDA and THCA synthase, compete for a common precursor, and copy number variation, as well as sequence variation at the loci encoding these enzymes has been proposed as a potential explanation for differences in ratios of the two compounds. However, according to Todd: ‘our understanding of the underlying genomic architecture of the CBDA and THCA synthase loci has been limited by the repetitive nature of the cannabis genome’.
To fully characterise the cannabis genome, including repetitive regions, the JCVI team performed whole genome sequencing using the long read nanopore technology. They chose a cannabis strain with high (15%) CBDA and low (0.3%) THCA. High-molecular weight DNA was extracted from young leaf tissue using a modified CTAB method. In total, the team generated 26 Gb of data, providing approximately 36x coverage of the 735 Mb genome. Following a genome assembly pipeline comprising minimap, miniasm, racon and pilon, and comparison to a genetic map, the genome could be resolved into 10 chromosomes.
Further analysis allowed the resolution of 14 CBDA and THCA synthase cassettes, 21 of which reside in two linked tandem arrays, nestled among a complex array of transposable elements on chromosome 9. Todd suggested that these transposons are potentially responsible for copy number variation of the synthase genes seen between different cultivars.
The team next utilised nanopore sequencing to generate full-length transcripts in order to support gene prediction and the identification of expressed synthase genes. Interestingly only one of the CBDA synthase gene loci is expressed. Todd stated that they have now observed this situation in two other high CBDA varieties.
The data also allowed them to clarify the lineage of the cultivar under investigation, which turned out to be a marijuana variety even though these varieties are traditionally classed as having very low levels of CBDA. Todd suggested that the CBDA in this variety is likely a recent introgression from hemp due to the breeders trying increase the levels of CBDA.
Wrapping up, Todd commented that using long-read nanopore sequencing, the team at JCVI have generated the most contiguous cannabis assembly to date, delivering new insights into the genetics of synthase genes and their evolution.






