Nanopore Community Meeting 2021: Day 2
With the Day 1 masterclass warm-up for the Nanopore Community Meeting 2021 complete, Day 2 saw over 4000 delegates from 113 countries begin to consume the latest updates from the Nanopore Community.
With three different content tracks in action (Human & Translational Research, Microbiology & Metagenomics and Plant & Animal genomics) delegates had a choice of topics available throughout the day. However, the conference started with a cross-track plenary talk on SARS-CoV-2 sequencing.
SARS-CoV-2 sequencing: on the midnight train to throughput - Julie Karl, Robert Maddox (University of Wisconsin-Madison, USA)
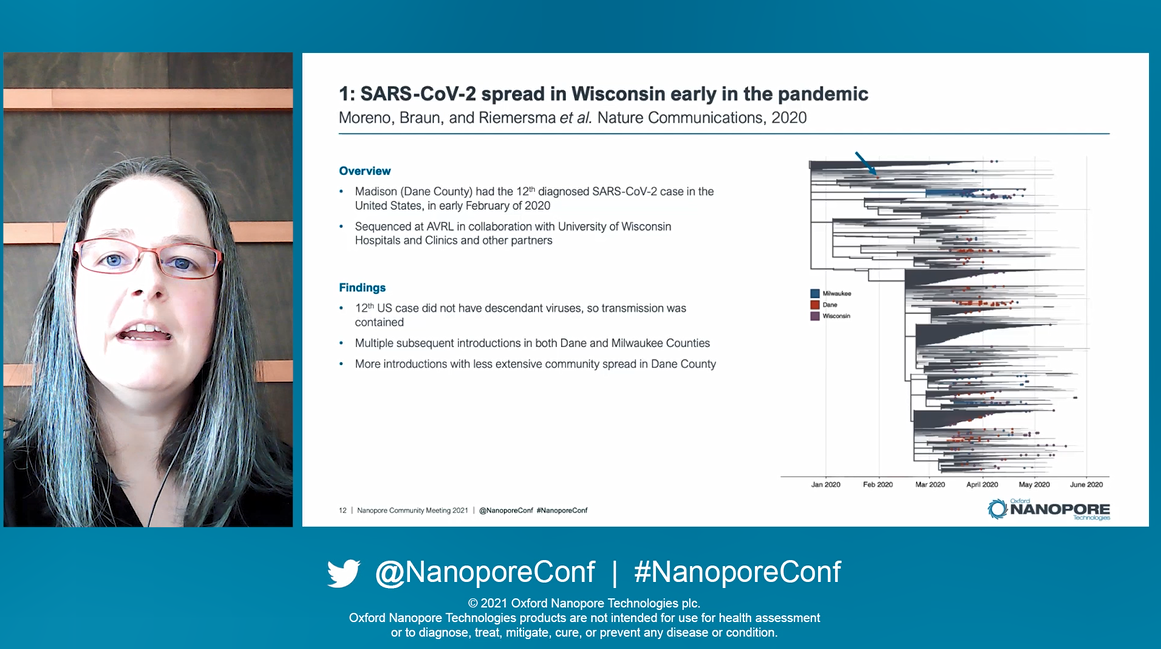 Julie and Robert discussed their team’s SARS-CoV-2 sequencing efforts in the state of Wisconsin, and their recent switch to the Midnight protocol. Julie began by presenting that on November 11th 2021, almost two years after the first SARS-CoV-2 case was detected, there were 251,266,207 confirmed cases worldwide and 5,070,244 deaths (as reported by the World Health Organisation). To combat this, a total of >7 billion vaccine doses had been administered. Despite this SARS-CoV-2 is still actively circulating with variants of concern arising. Ongoing, global and proactive study of the virus is evidenced by over 5 million SARS-CoV-2 genome sequences submitted to the GISAID database, ‘thanks largely to the ARTIC and ONT teams, for rapidly developing a protocol, deployable on such an easily accessible sequencing device’. Within Wisconsin specifically there have been >800,000 confirmed cases, of which ~3.7% have been sequenced. Julie's team, largely conducted of just two full-time employess, had themselves sequenced ~7.2% of positive cases within Milwaukee County, Wisonsin.
Julie and Robert discussed their team’s SARS-CoV-2 sequencing efforts in the state of Wisconsin, and their recent switch to the Midnight protocol. Julie began by presenting that on November 11th 2021, almost two years after the first SARS-CoV-2 case was detected, there were 251,266,207 confirmed cases worldwide and 5,070,244 deaths (as reported by the World Health Organisation). To combat this, a total of >7 billion vaccine doses had been administered. Despite this SARS-CoV-2 is still actively circulating with variants of concern arising. Ongoing, global and proactive study of the virus is evidenced by over 5 million SARS-CoV-2 genome sequences submitted to the GISAID database, ‘thanks largely to the ARTIC and ONT teams, for rapidly developing a protocol, deployable on such an easily accessible sequencing device’. Within Wisconsin specifically there have been >800,000 confirmed cases, of which ~3.7% have been sequenced. Julie's team, largely conducted of just two full-time employess, had themselves sequenced ~7.2% of positive cases within Milwaukee County, Wisonsin.
Through SARS-CoV-2 sequencing, Julie's team have been able to look at some interesting cases throughout the pandemic, including demonstrating the relationship between introductions of the virus into their country - and in one case, a patient who's transmission was compltely contained. Persistent SARS-CoV-2 infections in immunocompromised patients was another unique avenue they had the opportunity to explore, revealing receptor binding motif mutations observed between several immunocompromised patients. Her team have a contract with the CDC to continue sequencing and investigating SARS-CoV-2 in individuals with immune failure, such as infection despite vaccination, reinfection, and persistent infection, and plans to expand their work beyond SARS-CoV-2: investigating prolonged infection in the context of influenza. They are transitioning from the ARTIC to the Midnight protocol to improve throughput and decrease cost their of SARS-CoV-2 sequencing.
Robert's section of the talk provided an overview of real-world sample collection, including the challenges related to the sequencing of freeze-thawed samples. Demonstrating that they regularly exceed their sample number targets, he explained that, despite this, to achieve the required quality of consensus sequence for GISAID submission they must self-select for samples with lower Ct values. Due to their desire to sequence a range of Ct values, however, they are looking to develop strategies for sequencing a wide range of Ct values on a single flow cell.
One advantage Robert presented of the Midnight approach compared to ARTIC is that, due to the lower number of amplicons (29 vs. 98, respectively), future mutations in SARS-CoV-2 variants are less likely to fall in a primer binding site, and therefore less likely to impact sequencing. He pointed out how certain amplicons in the ARTICv3 protocol consistently underperformed, causing more frequent dropouts in the consensus; this wasn’t observed in the Midnight samples. They plan to further optimise the Midnight protocol; in particular, acknowledging that they will ‘always be working with potentially degraded samples’, and that there may be some primer optimisation they can make in line with their local variant pool.
For want of a nail: investigating somatic and germline variations in cancer - Sissel Juul, Phill James (Oxford Nanopore Technologies)
 Following Julie and Robert, Sissel Juul and Phill James from Oxford Nanopore Technologies took to the stage and presented work on eextrachromosomal DNA (ecDNA) in cancer, work completed by the Showcasing team from Oxford Nanopore. ecDNA, circular, double stranded and 1-3 Mb in length, is detected in up to 40% of solid tumours. Its presence is often associated with higher malignancy. They tend to contain highly expressed genes and can lead to tumour heterogeneity, and despite a desire to learn more about them, existing techniques often fall short: microscopy cannot inform on sequence; short reads cannot resolve structural variants; valuable methylation information is not obtained.
Following Julie and Robert, Sissel Juul and Phill James from Oxford Nanopore Technologies took to the stage and presented work on eextrachromosomal DNA (ecDNA) in cancer, work completed by the Showcasing team from Oxford Nanopore. ecDNA, circular, double stranded and 1-3 Mb in length, is detected in up to 40% of solid tumours. Its presence is often associated with higher malignancy. They tend to contain highly expressed genes and can lead to tumour heterogeneity, and despite a desire to learn more about them, existing techniques often fall short: microscopy cannot inform on sequence; short reads cannot resolve structural variants; valuable methylation information is not obtained.
Following sequencing of three neuroblastoma sell lines, Sissel's team managed to demonstrate that, potentially even with only a few hours of sequencing on a MinION flow cell, they could construct a circular ecDNA molecule from the CHP212 cell line. This had a high coverage across the MYCN oncogene locus, which they identified showed different MYCN sequences compared to the reference genome. They could also assess the methylation status of all three cell lines, and CHP212 demonstrated a region upstream of the MYCN locus which displayed a significantly different methylation profile. The significance of this is unknown, but it could relate to tumour heterogeneity observations.
Phill's team, looking at clinical applications, continued the cancer theme but instead with germline variants assessed using adaptive sampling. It's believed that 10% of cancers are hereitary, but Phil believes this to be an under-estimate: the impact of structural variants is probably insufficiently appreciated. Using adaptive sampling, a way of performing target enrichment on-flow cell using the MinKNOW software and requiring no additional library prep, he decided to investigate sequencing of a large panel of genes and chose Lynch syndrome as his muse. Following a quick Google to find the genes he wanted to target, and for good measure he included an expanded Lynch syndrome panel. Noticing that there were specific hereditary cancer gene panels he decided to add this, and following some more wide-spread additions he had generated a huge panel which would take months to years to analyse with a standard PCR-based approach. He decided to trial this with adaptive sampling.
An ovarian cancer sample, along with three cancer samples (one BRCA1/2 negative), were all sequenced for 72 hours on a GridION and he obtained 22.5 Gb per sample. Anticipating maybe 30x coverage for his chosen genes, they in fact obtained ~57x depth of coverage across the 201 cancer targets. Calling and assessement of SNPs identified a pathogenic SNP within in each cancer sample, whereas no SNPs were found in the a control. One of these was an intronic SNP within a breast cancer sample, to Phil a wonderful demonstration of why introns should be sequenced.
Tying together his talk Phil moved onto phasing and methylation, the former being particularly crucial in heterozygous disease conditions and the latter providing unexpected insights into regions such as the MSH2 gene promoter, within which all cancer samples showed increased methylation of a promoter-enhance region, but the control did not. This result wasn't expected, but was derived from the same native DNA dataset with no extra sample prep required. He closed on demonstrating that "rejected" reads from adaptive sampling could still be used to inform on copy number variation, before explaining the talk title: this comes from a proverb highlighting how small abberations can have large downstream effects.
Classification of pediatric acute leukemia using full-length transcriptomics - Jeremy Wang (University of North Carolina)
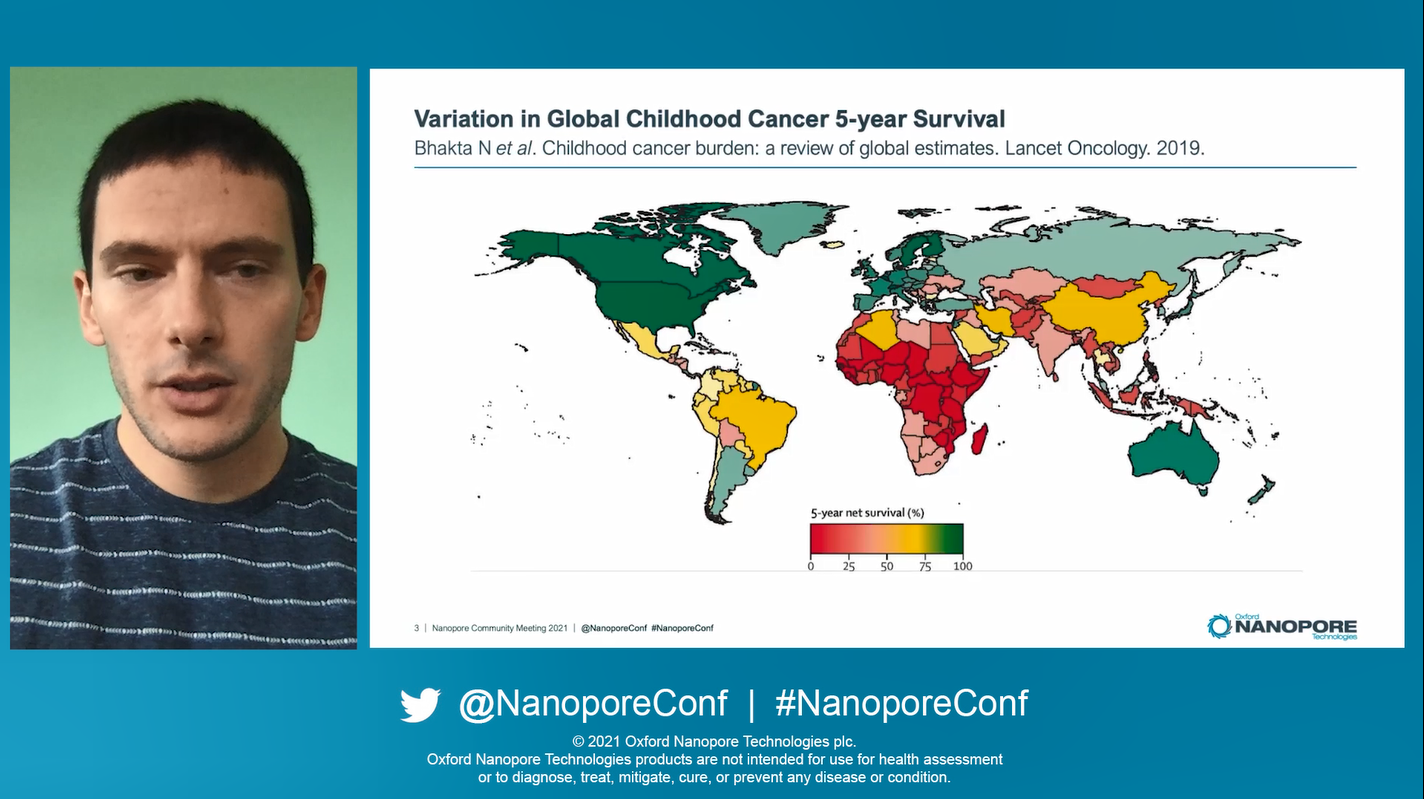 Entering our single track plenary sessions Jeremy Wang started the Human & Translational Research track off with his talk on pediatric acute leukemia. As a background to his work, there is a clear global discrepency in in childhood cancer 5-year survival rates, with a glaringly obvious pattern of poorer prognosis in lower income countries, includingparts of Africa, Asia, and America. This is underpinned by capital cost of equipment for characterising and diagnosis, particularly within genomics. Low-cost nanopore sequencing can leapfrog the barrier set by incumbent technologies.
Entering our single track plenary sessions Jeremy Wang started the Human & Translational Research track off with his talk on pediatric acute leukemia. As a background to his work, there is a clear global discrepency in in childhood cancer 5-year survival rates, with a glaringly obvious pattern of poorer prognosis in lower income countries, includingparts of Africa, Asia, and America. This is underpinned by capital cost of equipment for characterising and diagnosis, particularly within genomics. Low-cost nanopore sequencing can leapfrog the barrier set by incumbent technologies.
Gene expression profiling alone has been demonstrated able to classify paediatric leukaemia’s and their lineages, historically conducted with microarrays. This was an inflexible aproach, leaving much to be desired, and while short-read RNA sequencing is robust it carries significant capital costs. Jeremy set out to demonstrate that comparable classification of paediatric leukemias was possilble in low-resource settings using nanopore sequencing. This he demonstrated, even when using significantly degraded RNA —where many shorter reads exist, their sampling density across the transcriptome was comparable or higher than traditional short-read methods.
In order to achieve this he developed a number of machine learning and bioinformatics tools, combining partial least-squares (PLS) regression and a support vector machine (SVM) design to distinguish between expression profiles. Work with 211 nanopore-based transcriptomics profiles demonstrated promising results of almost 98% accuracy in aggregate, allowing him to define conserative cut-off values which accurately classify 95% of samples. For future work Jeremy plans to further improve this gene expression-based classificaiton, but importantly he is working with international collaborators in Malawi, Pakistan,and El Salvado to assess the feasibility of deploying his pipeline.
Resolution of complex human papillomavirus and human sequences - Nicole Rossi and Michael Dean (National Cancer Institute, USA)
 HPV causes 300,000 deaths world-wide per year. In order to study HPV and cervical cancer in low/middle-income countries, Nicole and her colleagues established a cohort in Guatemala and recruited over 700 women, collecting blood, tumour tissue, and clinical data. HPV integration can be quite complex and “there are often multiple copies of the HPV genome and flanking DNA generated during the integration process which are not resolved by short-read sequencing technologies”. The mechanism for these integration events is unknown but was investigated by Nicole using whole-genome sequencing andlong nanopore reads. Moreover, Nicole mentioned that adaptive sampling was used to target specific genes within the human and HPV genomes.
HPV causes 300,000 deaths world-wide per year. In order to study HPV and cervical cancer in low/middle-income countries, Nicole and her colleagues established a cohort in Guatemala and recruited over 700 women, collecting blood, tumour tissue, and clinical data. HPV integration can be quite complex and “there are often multiple copies of the HPV genome and flanking DNA generated during the integration process which are not resolved by short-read sequencing technologies”. The mechanism for these integration events is unknown but was investigated by Nicole using whole-genome sequencing andlong nanopore reads. Moreover, Nicole mentioned that adaptive sampling was used to target specific genes within the human and HPV genomes.
This approach allowed her to identify reads with human DNA on one end and HPV genomes on the other. Some of these HPV regions were inredibly long, with some HPV only reads of up to 160 kb. They see the same HPV genomes integrated in multiple chromosomes, and therefore deduced they must have originated before integration occurred —Nicole and her team have coined this phenomenon ‘superspreading’. A potential model for HPV superspreading is related to abnormal replication, leading to HPV concatemers being inserted at multiple loci in the human genome. Further to this however they identified that in some lines, not every tumour has an integrated virus - investigations into this identified a 150bp fragment of the E7 gene from HPV had inserted into an intron of the CEP126 gene, which was shown to lead to an increase of the YAP1 oncogene. Also demonstrated was the abilty for extrachromosomal episomal replication to cause cancer without integrating.
As a side note, Nicole added that using nanopore adaptive sampling, they could get a four-fold enrichment of HPV only containing reads, as well as HPV human reads. Some of the HPV containing reads were as long as 350 kb and human only reads as high as 1.5 Mb. These ultra-long reads helped identify blocks of HPV DNA flanked by human DNA.
Nicole then handed over to Michael, who documented his nanopore direct cDNA sequencing experiments to uncover full-length HPV transcripts in CaSki cell lines. How HPV expression is regulated, especially in absence of integration, is poorly understood however. In an effort to unravel just how expression is controlled, Michael sought to examine the epigenetic profile of the cell lines and tumours. 5-methylcytosine (5mC) was called from direct nanopore sequencing using Megalodon.Michael justified his choice to use nanopore sequencing by stating “the attraction of the nanopore data is that for the reads across the full HPV genome we can see the phase of all the methylated bases and no other technology can show this”.
Further work will focus on the observation that integration activities can disrupt important cellular genes and oncogenes, and how this can occur. To that end, Pore-C, a chromatin interaction method adapted to nanopore sequencing, was used. In doing so, Michael and his teamhave 8 million reads, with over 3 million contacts, of which over 1 million occur at long range. Interestingly, there are over 12,000 HPV-containing reads in the dataset and 8,000 of theseconnect to human sequences. Michaelhopes that this will shed light on how HPV integration influences the human genome.
Michael closed by presenting data on the PI3K signalling pathway, in which almost of cervical tumours have abberations. Michael proceeded to test the drug Piqray, a PI3KCA inhibitor used for treatment of breast cancer. Several cervical cancer cell lines revealed a marked reduction in expression of key HPV genes, as well as dramatically reducing proliferation: promising findings which Michael plans to follow up by investigating how methylation and chromatin structure respond to treatment with the drug.
Genomic skimming on the MinION uncovers cryptic hybridization in the buffy-tufted marmoset, one of the world’s most threatened primates - Joanna Malukiewicz (German Primate Center, Germany)
 With a switch from the human focus, Joanna Malukiewicz took us through her work on the buffy-tufted marmoset.
With a switch from the human focus, Joanna Malukiewicz took us through her work on the buffy-tufted marmoset.
There are only 10-11k adult buffy-tufted marmosets (Callithrix aurita) estimated remaining in the wild in southeastern Brazil, lead to due to a loss of habitat & disease. A small primate weighing ~400 g, it is listed on the International Union for Conservation of Nature (IUCN) Red List, there are multiple ecological and anthropogenic threats to them. Illegal trafficking of other marmoset species results in ecological competition, and infectious diseases such as Yellow Fever result in decreased opportunities for mating. More concerning however is the inter-species breeding between C. aurita, C. jacchus and C. penicillata. The relativey young evolutionary age between these species means they are able to interbreed, resulting in offspring from interspecific hybridisation which tend to have a ‘koala bear’ appearance.
C. auritais remains relatively understudied. Therefore, Joanna and her team setup a goal to ‘develop a rapid, cost-effective protocol to quickly obtain genomic and genetic data for Callithrix aurita’, in order to provide data critical to the conservation of the species. To this end, they sampled DNA from an individual with a C. aurita phenotype and assessed whether they could use nanopore sequencing on a MinION device to reliably reconstruct its mitogenome for use in phylogenetic and genetic diversity analyses.
Low-coverage nanopore sequencing of gDNA from individuals with a C. aurita phenotype on 2 MinION Flow Cells, combined with genome skimming for mtDNA allowed them to construct a full mitogenome from ~9x average genome coverage depth. Using a consensus between this and a Sanger-sequencing-generated mitogenome, they then constructed a robust maximum-likelihood phylogeny. The team were expecting for their sequenced individual to group within the C. aurita clade; however, its haplotype in fact grouped within a C. penicillata clade. Among others, this result, Joanna highlighted, represented the ‘first published accounts of introgression of genetic material from exotic C. penicillata marmosets into C. aurita’.
Turning to the practicalities of the approach, Joanna was happy to say that it 'really holds great potential for enhancing mitogenomics and genetic diversity studies of data-deficient and/or non-model organisms’. Sample prep was less than one day, and sequencing took 48 hours. Developments in sample preparation methods also mean less input material would now be required.
Joanna hopes that that larger sampling efforts can now be undertaken to allow full understanding of the genetic diversity present - her method, combined with the MinION for in-field sequencing, could make this acutely possible.
ORG.one: a new program to promote sequencing biodiversity - Tomas Marques-Bonet (Institute of Evolutionary Biology, Spain)
 Bringing to a close the Community plenaries for the day, Tomas Marques-Bonet started with the impactful statement that we are in the 6th mass extinction of species but crucially (sadly?), 'this time it is because of us.' The International Union for Conservation of Nature (IUCN) has declared that there are tens of thousands of species currently threatened with extinction; this is 28% of all assessed species (including both plants and animals), with is a particular threat for amphibians.
Bringing to a close the Community plenaries for the day, Tomas Marques-Bonet started with the impactful statement that we are in the 6th mass extinction of species but crucially (sadly?), 'this time it is because of us.' The International Union for Conservation of Nature (IUCN) has declared that there are tens of thousands of species currently threatened with extinction; this is 28% of all assessed species (including both plants and animals), with is a particular threat for amphibians.
There is no easy solution for this or the problem would already have been solved. Action to resolve the situation, Tomas was keen to present, has to be taken from economic, social, ecological and topically, genetic perspectives. Whilst genetics, in particular, isn't the sole solution, geneticists have a lot to contribute when it comes to take steps to solve the problem - you need to be able to quantify the issue - how healthy a population is, for instance - before you can begin to take steps to prevent the dying out.
With Tomas' own experience in great ape genomics, he used this as an example of how a combination of high-quality reference data and knowledge can become key for decision making for the protection of the species. Thankfully a number of global initiatives are taking steps to capture a snapshot of global diversity, with Oxford Nanopore itself being responsible for the ORG.one initiative. This has already begun to get underway with nine species selected including birds, mammals, and amphibians. Despite the samples for these species having been in storage, high-quality genomes are already being generated which has been largely aided by the impressive read engths obtained off the samples. Read of N50 of ~20–40 kbps, which is ‘really good’, lead in turn to impressive assembled genomes, with contig N50s of ~30–50 Mbps, ‘which is quite remarkable’. He used examples from the spider monkey, the blue-throated macaw and the Montseny brook newt to demonstrate this, the latter being the most endangered amphibian in Europe.
No reference genome exists for this newt, partly due to its 'colossal genome' full of repeats and redundancies which, of course, long nanopore reads can help to solve. Currently a genome coverage depth of ~20x has been obtained with nanopore sequencing from one individual, and the data allows them to do 'a lot of beautiful stuff'. An example given is that with the collaboration of another team they hope to generate data fround around 200 newts, helping to further uncover more about the genetic diversity of this species.
To close, Tomas highlighted that all the data being produced for this project is online and open-source. He hopes the community is excited and engaged leading to more studies in the future which help to protect the fragile diversity of our natural world.
Finishing up Day 2 were two talks from Oxford Nanopore on their newest technology updates. Read more about it here.






