Nanopore sequencing and the Arabidopsis centromere paradox
The centromere paradox
The Arabidopsis genome was first sequenced over 20 years ago — yet the centromeres, due to their highly repetitive nature, have remained an enigma until only recently. Using long, PCR-free nanopore reads, Ian and his team have been able to sequence Arabidopsis thaliana centromeres and resolve their structure. He had expected that all five centromeres in Arabidopsis would be very similar, but quickly saw that they all had their own ‘flavour’ of the satellite repeat, CEN180. He noted how, though all comprised of the same repeat, ‘in terms of exact sequences, centromere 1 had its own subfamily – and similar patterns are also seen in human centromeres. That was quite unexpected’.
‘There are a lot of mysterious aspects to the centromeres… there’s remarkable diversity… the centromere is, even between very closely related species, sometimes unrecognisable at the sequence level.'
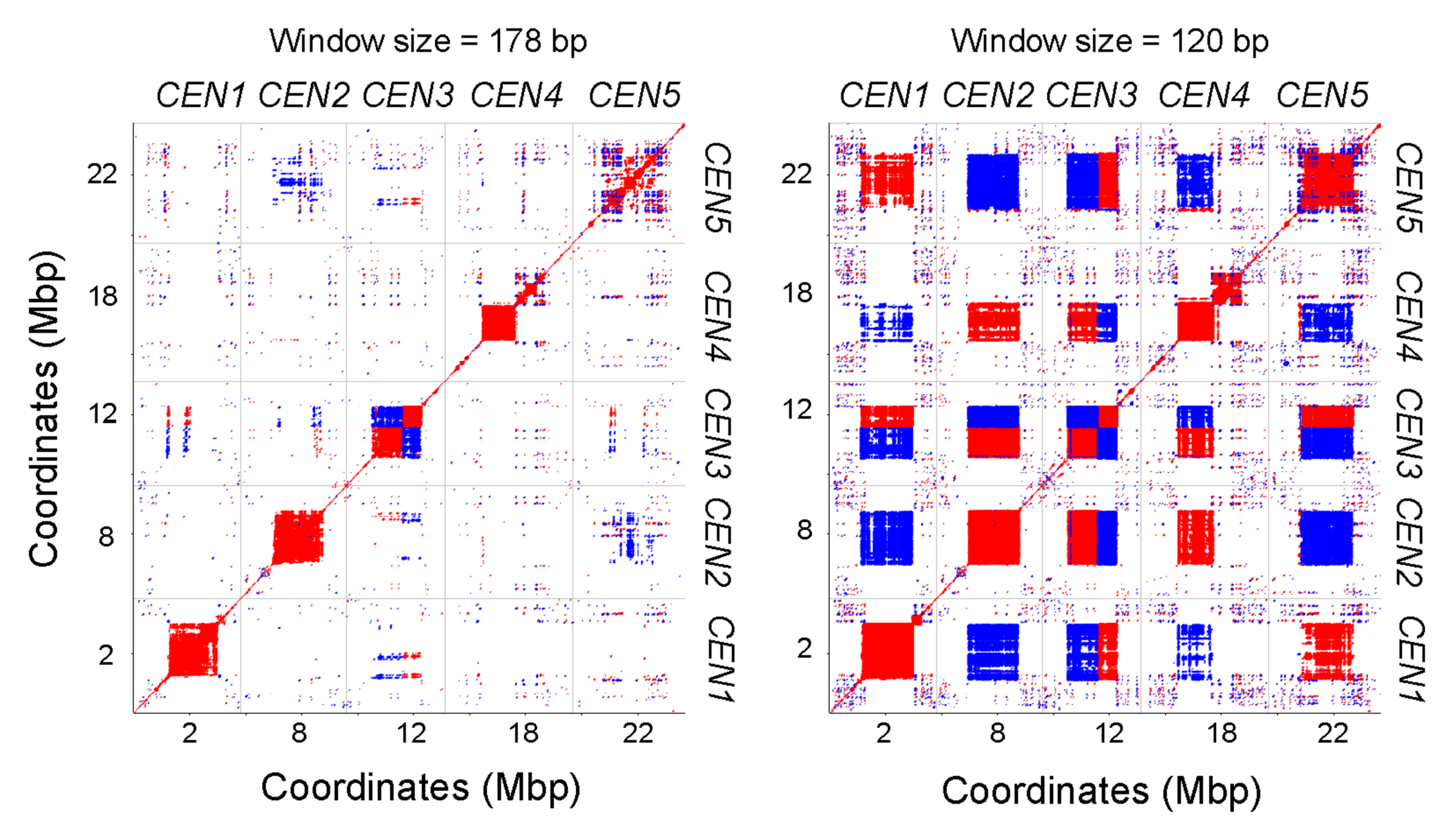
Figure 1: Dot plots comparing the five completely assembled Arabidopsis centromeres.
Red and blue indicate forward- and reverse-strand similarity, respectively. From Naish et al. (2021).
Their next surprise concerned the presence of a type of retrotransposon known as ‘Athila’. Though known to feature in Arabidopsis, they had generally been considered as a ‘dead’ or ‘degraded’ family; however, Athila found in the centromeres were found to be unexpectedly intact and young: ‘the living ones were buried inside, deep in these parts of the genome. It's unclear why they are adapted to integrate in the centromeres.'
Centromeres exist in each chromosome, and their role is to enable a complex of proteins – the kinetochore — to assemble during mitosis and meiosis, so that chromosomes can be pulled to opposite poles of dividing cells. Considering that the centromere is central to a highly conserved function, Ian explained, it’s paradoxical that it is one of the fastest evolving and most diverse parts of the genome. What could be behind this? 'My gut feeling is that the Athila might be something to do with this. Transposons generally are thought to behave selfishly'. Ian described the phenomenon of centromere ‘drive’, in which the centromere can also act as a ‘selfish’ element to increase its own transmission during cell division. This can result in very unequal, non-Mendelian transmission of centromeres — Ian noted an example in monkeyflower, where this uneven inheritance reaches 98%. Ian explained, ‘if the Athila could influence centromere function during cell division, they could try to bias the system to their own advantage. To try and counteract that, one defence may be constantly re-writing everything to get rid of these elements'.
Long repeats meet long reads
The full repeat array Ian and his team have seen in Arabidopsis centromeres is between 2–4 megabases in length. Remarkably, this makes them comparable in length to human centromeres, despite their overall chromosome sizes being a fraction of the length; as it stands, it isn’t clear why this might be. Ian stressed the significance of long nanopore reads in assembling such regions. Whilst repeat polymorphism and Athila retrotransposons can aid centromere assembly by providing ‘landmarks’, Ian describes how ribosomal RNA (rRNA) is more difficult still, with repeat units reaching kilobases – ‘which is why Nanopore…is the only way you can get through ribosomal DNA currently’.

Figure 2: Dot plot of centromeric ATHILA retrotransposons. Red and blue
indicate forward- and reverse-strand similarity, respectively. From Naish et al. (2021).
‘I think eventually with ultra-long-read sequencing all genome regions will be accessible — but there are some very repetitive genomes out there, so challenges remain.’
Exploring epigenetics
Ian’s group are also using nanopore technology to investigate the potential role of epigenetic modifications in A. thaliana centromeres. Previously, Ian’s team had to perform bisulfite conversion followed by short-read sequencing, but now, with PCR-free nanopore sequencing, base modifications can be detected directly in native DNA strands: ‘you don’t have to convert anything — which makes mapping , with the long reads in repeat regions much better than with short reads. It’s like night and day’.
Ian and his colleagues are especially interested in is investigating methylation in non-CG contexts, such as CHH. Ian explained that CG methylation in plants is very dependent on the enzyme MET1, whereas non-CG methylation has a different maintenance pathway. If you knock out MET1, most of the non-CG methylation remains intact in the chromosome arms, as expected; however, Ian revealed that ‘that’s not true in the centromeres’: the non-CG methylation sites are unmethylated in MET1 knockouts. He described how ‘that was unexpected.... we don’t understand it, but it’s an important finding. Without having an assembly and without being able to look at the methyl state, we wouldn't know any of this'.
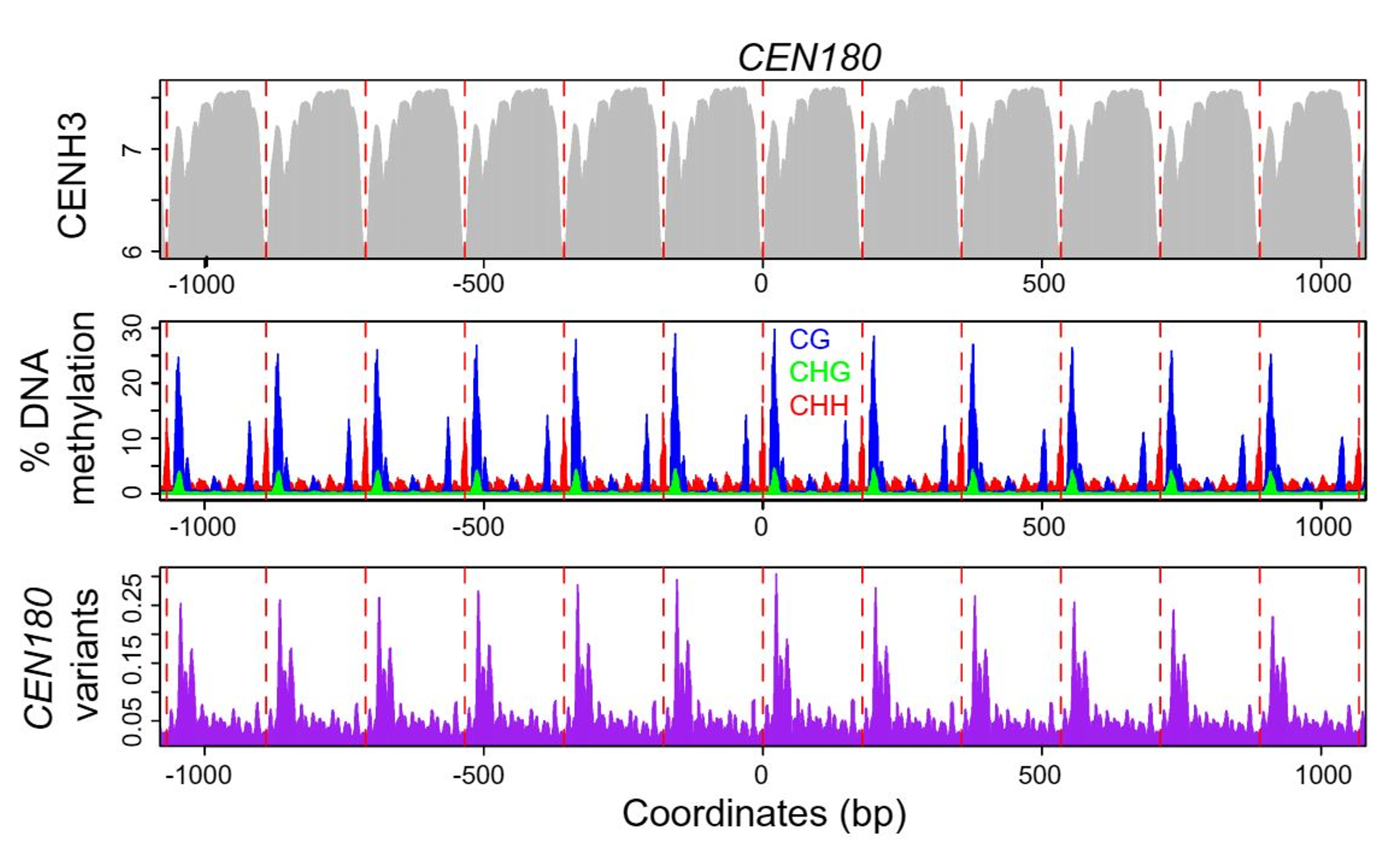
Figure 3: plots of CENH3 histone variant ChIP enrichment (grey), DNA methylation in CG (blue), CHG
(green), and CHH (red) contexts, and CEN180 satellite variants (purple), averaged over windows centered on
CEN180 starts. The red dashed lines show 178 bp increments. From Naish et al. (2021).
Using Pore-C — Oxford Nanopore’s workflow combining chromatin conformation capture with long nanopore reads — Ian is also hoping to investigate not just higher order structure, but also long-range, phased methylation: ‘what we’re really excited about at the moment is, because the reads are so long, you’ve now got information on adjacent [methylation] sites that you just never had from short-read bisulfite. Having these haplotypes of methylation is a game changer’.
‘There are just so many possible applications to the technology.’
Ian and his team also like to experiment and combine techniques, such as coupling chromatin immunoprecipitation (ChIP) with nanopore technology. Ian explained that ‘in any system which has methylation, people would be really interested to know...'is my transcription factor binding to unmethylated sequences or methylated?'’. Describing how they sequenced a ChIP sample on a Flongle, Ian highlighted how ‘the really amazing thing was that we didn’t do any amplification, so you can see the methyl state... of the ChIP fragments, which with [short-read sequencing] and those approaches, all that information just gets lost during amplification’.
Revisiting old questions
Going back to the beginning of his time using nanopore sequencing, Ian noted that, with one or two MinION Starter Packs, they were able to start sequencing in their own lab. He described how, whilst traditional sequencing methods may require samples to be sent to a sequencing facility, with higher costs and turnaround times of a couple of weeks, ‘one of the really nice things about nanopore is that nearly anyone can afford to just have a go at it and just see whether it works for them’.
‘The thing that really got us hooked was how accessible the technology is… you can just do it easily and quite cheaply in your own lab.’
What’s next for Ian’s research? He plans to investigate centromere diversity — and what could be driving it — further. He points out how such repetitive sequences were, until relatively recently, considered ‘junk DNA’ — but that he thinks it’s anything but that. ‘Junk DNA, selfish DNA — there’s lots of mysterious elements to that. Suddenly we have access to a lot more information about it. It’s a good time to revisit those questions. I think it’s going to be exciting, the next couple of years, for the whole field’.
Ian stressed that, whilst crop plant genomes are widely studied, others are much less well characterised – and there are many plants that Ian would like to sequence. Paris japonica at 149 billion base pairs (50x larger than the human genome) is high on his list: ‘my neighbour had a Paris japonica and I used to look at it thinking ‘I really want to assemble that''. Also written on his list of grant application ideas is, simply, ‘trees?’. Ian asked: if you were to sequence samples from an oak tree that was several hundred years old and compare methylation from branches on opposite sides, ‘would it have stayed the same over those hundred years? Or do the different branches of the tree have different genomes?’ If you do get round to that experiment Ian, please let us know.
Want to learn more?
Watch Ian’s talk on The genetic and epigenetic landscape of the Arabidopsis centromeres at the NCM 2021
View our White paper on Closing the gap in plant genomes
Find out more about plant research with nanopore sequencing
Images from Naish et al. (2021) reproduced with permission from Science. Readers may view, browse, and/or download material for temporary copying purposes only, provided these uses are for noncommercial personal purposes. Except as provided by law, this material may not be further reproduced, distributed, transmitted, modified, adapted, performed, displayed, published, or sold in whole or in part, without prior written permission from the publisher.
1. Naish, M. et al. The genetic and epigenetic landscape of the Arabidopsis centromeres. Science. 374(6569) (2021). DOI: 10.1126/science.abi7489






