Delivering ‘unique insights’ into infection with direct RNA sequencing
)
- Published on:
Full characterisation of influenza virus in a single assay
Influenza is a highly contagious respiratory illness caused by influenza viruses. These relatively small RNA viruses (genome size: ~13.5 kb) are exceptionally adaptable and efficient at evading the host immune response, making producing a long-lasting vaccine against them challenging.
Over the years, efforts to study RNA viruses have been limited by the available techniques. Legacy sequencing technologies can only read short fragments of RNA, necessitating the use of often inexact computational assembly methods. In addition, the requirement for cDNA conversion not only introduces potential amplification bias but also overwrites base modifications, hiding potentially important epitranscriptomic insights. Now, using nanopore sequencing, native RNA can be directly sequenced, quantified, and analysed for base modifications, all in a single assay.
Wang et al., from the University of Oklahoma, USA, used nanopore direct RNA sequencing to explore the changes in viral and host transcripts, and the host immune response, during influenza infection1. Using the Direct RNA Sequencing Kit and benchtop MinION sequencing device, the team analysed mRNA transcripts and non-coding RNA (both polyadenylated and non-polyadenylated) from influenza-exposed and mock-exposed human bronchial epithelial research samples. In a single assay, they were able to measure base modifications, transcript isoforms, poly-A tail length, and differential expression.
Generating novel insights
Differential transcript expression analysis suggested the influenza-exposed samples were at the early stages of infection, since the most significantly upregulated and downregulated transcripts related to immune response and virus entry. This was further supported by the lack of viral RNA transcripts detected in the bronchial samples — consistent with the early stages of infection when the virus is bound to cells but has not yet entered or begun to replicate.
Direct detection of base modifications revealed 502 transcripts with m6A methylation in influenza-exposed samples, and 395 transcripts with m6A methylation in mock-exposed samples; however, the abundance of the majority of transcripts exhibiting differential m6A did not change. The team suggested that, although m6A methylation is an important regulatory mechanism of the transcriptional response to viral infection, it is unlikely to be the main mechanism regulating transcriptional changes in response to influenza in their model system.
The researchers further discovered that m6A methylation did not correlate with alternative splicing of mRNA in influenza-exposed samples. Unambiguous identification of complex, alternatively spliced isoforms is possible using nanopore sequencing as entire strands of native RNA are read in a single continuous read.
Of note, the team identified a significant increase in expression of one of the eight transcript isoforms of stimulated gene 12 (ISG12). This gene is known to suppress viral infection and exhibits complex alternative splicing. The researchers highlighted that ‘such complex alternative splicing is difficult to identify in short-read sequencing with data from reads 100–200 bp in length’.
‘nanopore data unambiguously identifies the isoforms of transcripts that have multiple alternative splice sites’1
Methylation did not correlate with poly-A tail length either. An abundance of genes (410) associated with immunity and immune regulation showed a change in poly-A tail length. However, those with the greatest change in gene expression were less likely to show a change in poly-A tail length. This led the team to hypothesise that there are distinct, independent mechanisms of transcriptional and post-transcriptional regulation of host response genes during influenza infection.
Interestingly, the team observed increased methylation in response to influenza exposure in two long non-coding RNAs (lncRNAs) — CHASERR and LEADR, which were recently discovered to have roles in the immune response. Explaining that increased methylation could change the shape of the lncRNA, they predict methylation may disrupt the molecular interactions through which these lncRNA perform regulatory functions. In addition, the expression levels of several small nucleolar RNAs (snoRNAs), a type of non-coding RNA, were decreased in response to influenza. The snoRNAs guide pseudouridylation. Nanopore sequencing revealed a corresponding decrease in pseudouridylation in two novel lncRNAs.
Although further research is necessary to fully understand the mechanisms through which noncoding RNA and RNA modifications fine tune the host response to influenza infections, the team highlighted how direct RNA sequencing using nanopore technology revealed ‘unique insights’.
‘The ability to identify multiple modifications on a long read of an individual RNA molecule surpasses other techniques’1
Evaluating the latest direct RNA sequencing chemistry
Furthermore, Hewel et al.2 evaluated the latest nanopore RNA chemistry and basecalling model integrated into MinKNOW, the software onboard nanopore sequencing devices. Using a PromethION device, they sequenced standardised RNA test samples from HEK293T and Universal Human Reference RNA (UHRR) cell lines, and human blood research samples. Reporting increased sequence accuracy and throughput, the team also highlighted the ability to detect native RNA modifications ‘in a close to real time’ (Figure 1). The researchers shared how the new chemistry opens up ‘very intriguing possibilities’, including the potential for RNA diagnostics, and possible future clinical applications observing RNA modification disorders as a routine measure.
‘The new chemistry has significantly improved throughput and accuracy and can support real-time tracking of state-specific methylation information’2
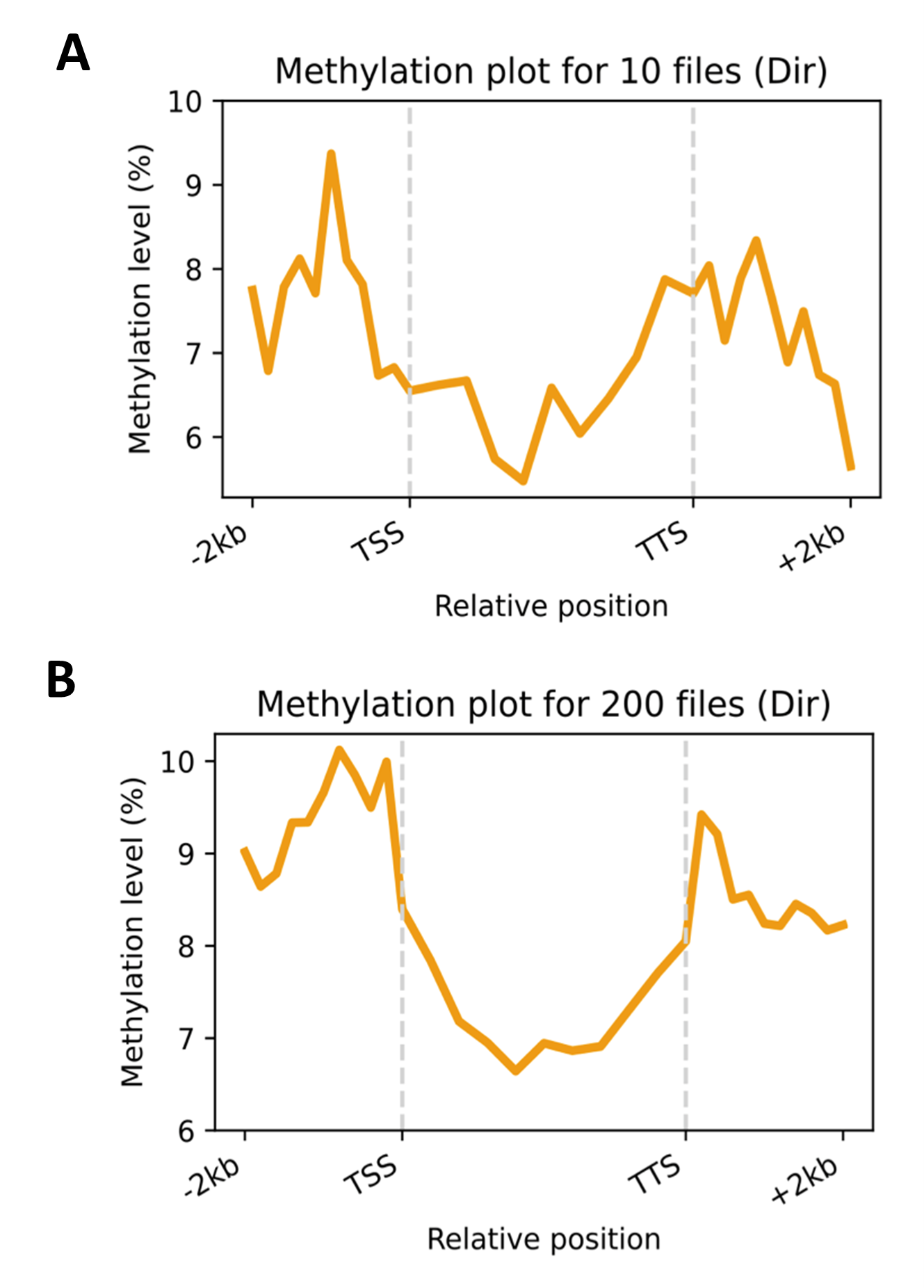
Figure 1. Direct (dir) detection of m6A in blood research samples for sequences up to 2 kb upstream of the transcription start site (TSS), gene bodies, and sequences up to 2 kb downstream of the transcription termination site (TTS). Plots were created in near real time after every a) 10 files and b) 200 files output by MinKNOW. Figure taken from Hewel et al.2 and made available under Creative Commons License (creativecommons.org/licenses/by-nd/4.0).
This case study was taken from the RNA sequencing white paper.
Wang, D. et al. Nanopore direct RNA sequencing reveals virus-induced changes in the transcriptional landscape in human bronchial epithelial cells. bioRxiv 600852 (2024). DOI: https://doi.org/10.1101/2024.06.26.600852
Hewel, C. et al. Direct RNA sequencing (RNA004) allows for improved transcriptome assessment and near real-time tracking of methylation for medical applications. bioRxiv 605188 (2024). DOI: https://doi.org/10.1101/2024.07.25.605188






