Adeno-associated virus (AAV) sequencing - development notes and best practices

Requirements
Adeno-associated virus (AAV) sequencing - development notes and best practices
FOR RESEARCH USE ONLY.
Contents
Introduction
Preparation prior to AAV sequencing
Results
- 1. Extraction methods
- 2. Annealing vs direct ligation
- 3. Rapid barcoding DNA V14 (SQK-RBK114.24 or SQK-RBK114.96) library preparation
Notes on downstream analysis of AAV
Summary
Introduction
In our current protocol for sequencing adeno-associated viruses (AAV), Adeno-associated virus (AAV) sequencing from recombinant adeno-associated virus (rAAV) vectors using SQK-NBD114.24, the Native Barcoding Kit 24 V14 (SQK-NBD114.24) is used to sequence up to six samples. Below, we explain the alterations that you can make to optimise the protocol for your experiment, including our evaluation of alternative extraction methods and annealing steps on AAV genomic material.
AAV structure definitions:
Single stranded AAV (ssAAV) only has alignments to one strand
Self complement AAV (scAAV) has alignments to both strands. If there are more than two alignments for a read, the structure is defined as ‘complex’.
Partial ssAAV can be either:
- Incomplete genome (ICG): one alignment that does not reach the full ITR-ITR length of the reference.
- Genome deletion mutant (GDM): there are two alignments to the same strand but with a gap between alignments.
Note: If a read contains only part of the ITR, either with one or two alignments to an ITR region, then it is classed separately from the ssAAV and scAAV.
Preparation prior to AAV sequencing
Prior to extraction and sequencing of AAV, we recommend sequencing the constructed recombinant AAV (rAAV) vectors, as well as any additional helper and repcap plasmids used for transfection and production of AAV preparations. We used the Rapid sequencing V14 - Plasmid sequencing (SQK-RBK114.24 or SQK-RBK114.96) protocol to prepare and sequence AAV plasmids and the wf-clone-validation workflow available from EPI2ME to assemble both recombinant AAV preps (Figure 1.) and helper/repcap plasmids (data not shown). Sequencing of AAV plasmids can be used to confirm plasmid constructs, reducing the likelihood of truncations and contaminating AAV genomes in final preparations.
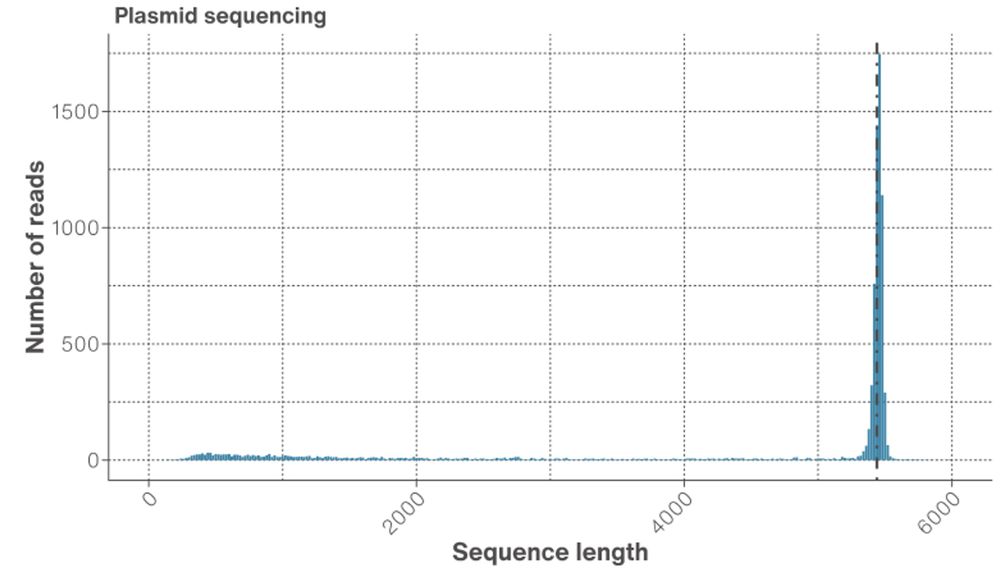 Figure 1. Read length distribution. The N50 is represented by the dotted line at the peak, which is representative of full length plasmids (~5.3 kb).
Figure 1. Read length distribution. The N50 is represented by the dotted line at the peak, which is representative of full length plasmids (~5.3 kb).
Results
Extraction methods
Extraction methods were tested using an AAV8 serotype sample containing a truncation in the genome to identify the method that preserves the most long reads and full-length ITR-ITR genomes. The AAV8 sample was prepared using the following methods:
- Heat at 95°C
- Proteinase K treatment
- Heat at 95°C and proteinase K treatment
- Phenol:chloroform-based method
- PureLink™ Viral RNA/DNA Mini Kit extraction protocol
After extraction, we prepared the samples using the Ligation Sequencing Kit V14 (SQK-LSK114) and sequenced for ~19 hours with HAC basecalling and the minimum read length set to 20 bp. All methods resulted in both an increase in short reads and a decrease in full length ITR-ITR genomes (~4.2 kb), in comparison to the PureLink™ Viral RNA/DNA Mini Kit method.
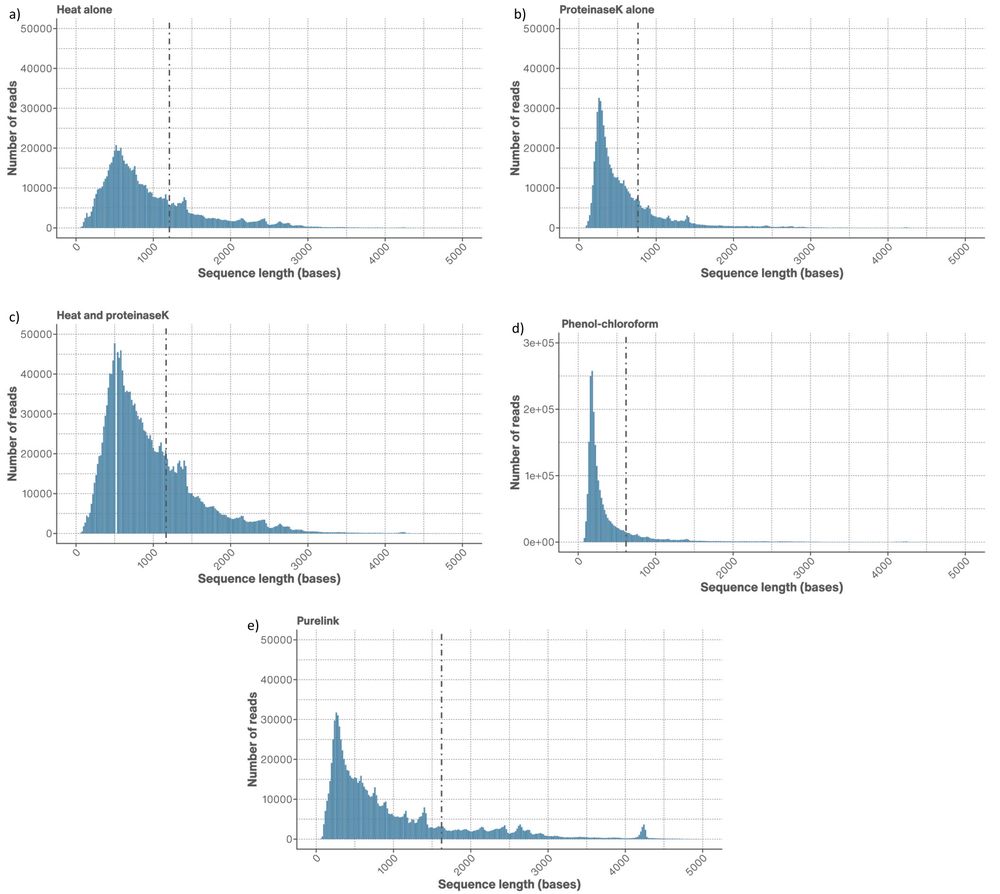 Figure 2. Read length distributions of the truncated AAV genomes extracted using a) heat only, b) proteinase K only, c) heat and proteinase K, d) phenol-chloroform, and e) PureLink™ Viral RNA/DNA Mini Kit extraction. The dotted line represents the N50 on each extraction plot and passed reads (Q-score ≥9 were used only).
Figure 2. Read length distributions of the truncated AAV genomes extracted using a) heat only, b) proteinase K only, c) heat and proteinase K, d) phenol-chloroform, and e) PureLink™ Viral RNA/DNA Mini Kit extraction. The dotted line represents the N50 on each extraction plot and passed reads (Q-score ≥9 were used only).
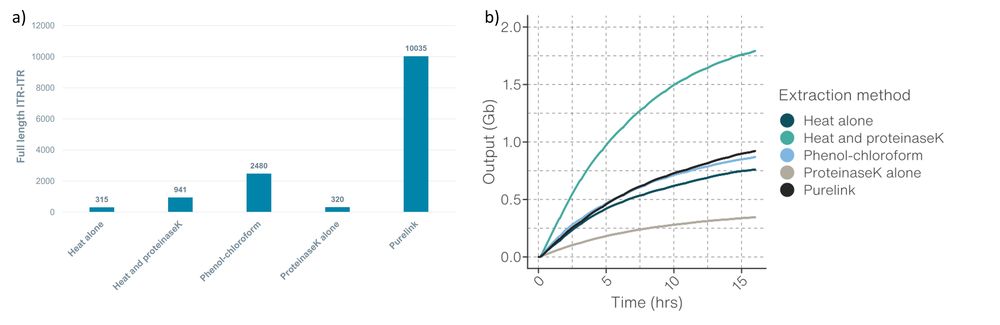 Figure 3. a) Number of full-length ITR-ITR reads for each extraction method. b) Data output for each extraction method.
Figure 3. a) Number of full-length ITR-ITR reads for each extraction method. b) Data output for each extraction method.
Annealing vs direct ligation
Annealing steps are often used in AAV sequencing protocols to anneal the two (+) and (-) genome strands of extracted single stranded AAV (ssAAV) prior to library preparation. The effects of two annealing protocols on either ssAAV or a mixture of ssAAV and scAAV prior to ligation were compared with a matched sample that was not annealed and directly ligated in the library preparation.
The annealing methods:
- High temperature annealing: AAV8 serotype extracts (in 50 mM NaCl and 5 mM Tris-HCl, final pH 7.5) were heated for 5 minutes at 95°C and then slow ramping down to 25°C (ramping rate: -1°C/min).
- Lower temperature annealing: AAV8 serotype extracts were separately alkaline denatured with 0.2 N NaOH, before being neutralised using Tris-HCl (pH 8.0), ethanol precipitated and resuspended in 1x SSC buffer. AAV genomic material was then heated to 70°C before cooling to 20°C (ramping rate: -1°C/min).
Three AAV extracts were taken into library preparation using the Ligation Sequencing Kit V14 (SQK-LSK114). Two of the AAV extracts were annealed using the high temperature and low temperature method. The third extract was not annealed and directly ligated in the library prep. This extract had a faster library prep time and more full-length ssAAV compared to the annealed extracts (Figure 4.). All AAV sequencing was carried out on individual MinION flow cells for 72 hours with minimum read length reduced to 20 bp and using the High-accuracy (HAC) live basecaller.
Femtopulse traces were generated for all extracts prior to library prep and after end-prep to further demonstrate the effects of the annealing methods on the AAV genome.
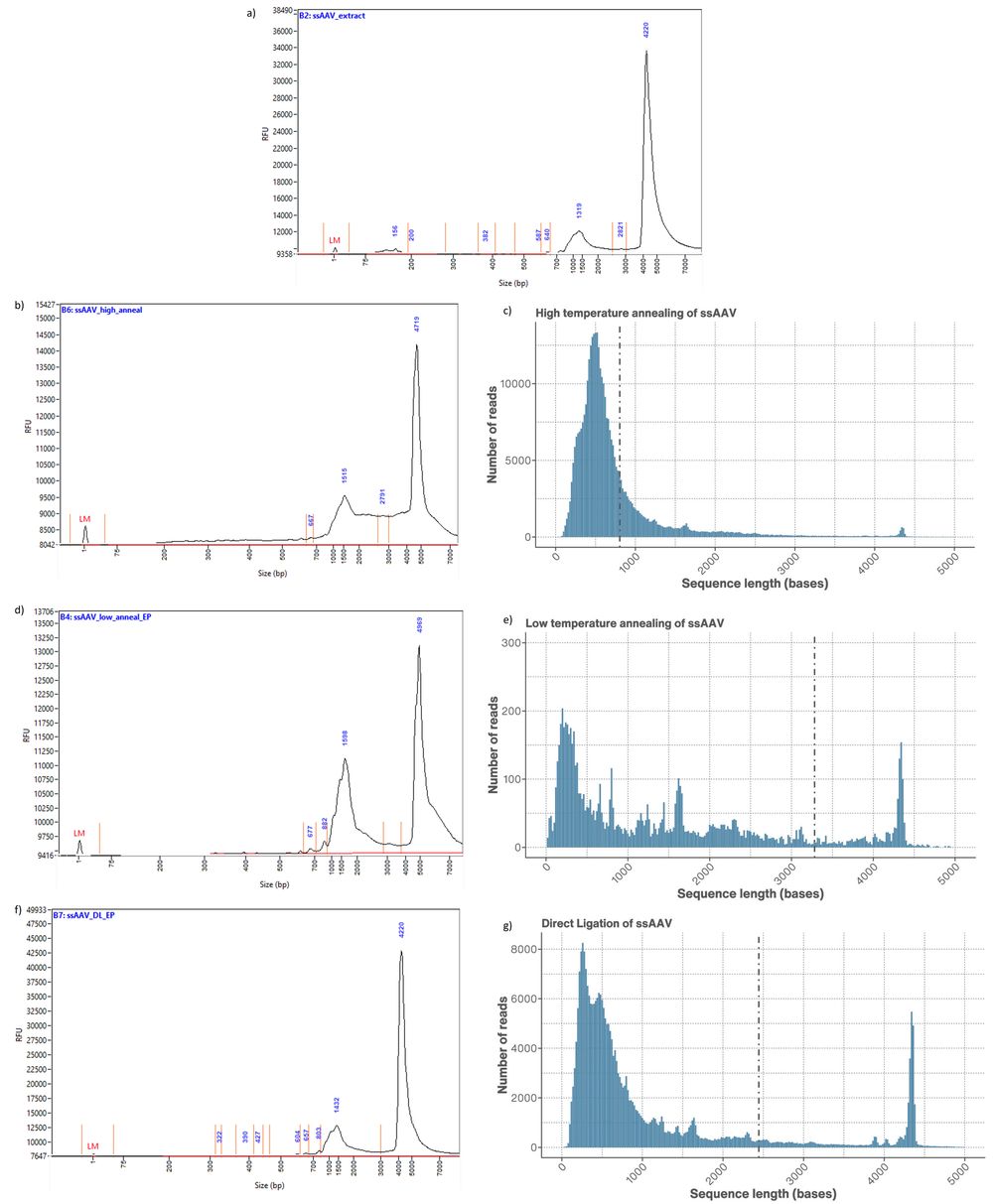 Figure 4. a) Femtopulse of ssAAV present in the initial AAV extract trace. b) Femtopulse trace of ssAAV annealed using the high temperature method compared to c) the read length distribution of the same ssAAV sample. d) Femtopulse trace of ssAAV annealed using the low temperature method compared to e) the read length distribution of the same ssAAV sample. f) Femtopulse trace of ssAAV annealed using direct ligation compared to g) the read length distribution of the same ssAAV sample.
Figure 4. a) Femtopulse of ssAAV present in the initial AAV extract trace. b) Femtopulse trace of ssAAV annealed using the high temperature method compared to c) the read length distribution of the same ssAAV sample. d) Femtopulse trace of ssAAV annealed using the low temperature method compared to e) the read length distribution of the same ssAAV sample. f) Femtopulse trace of ssAAV annealed using direct ligation compared to g) the read length distribution of the same ssAAV sample.
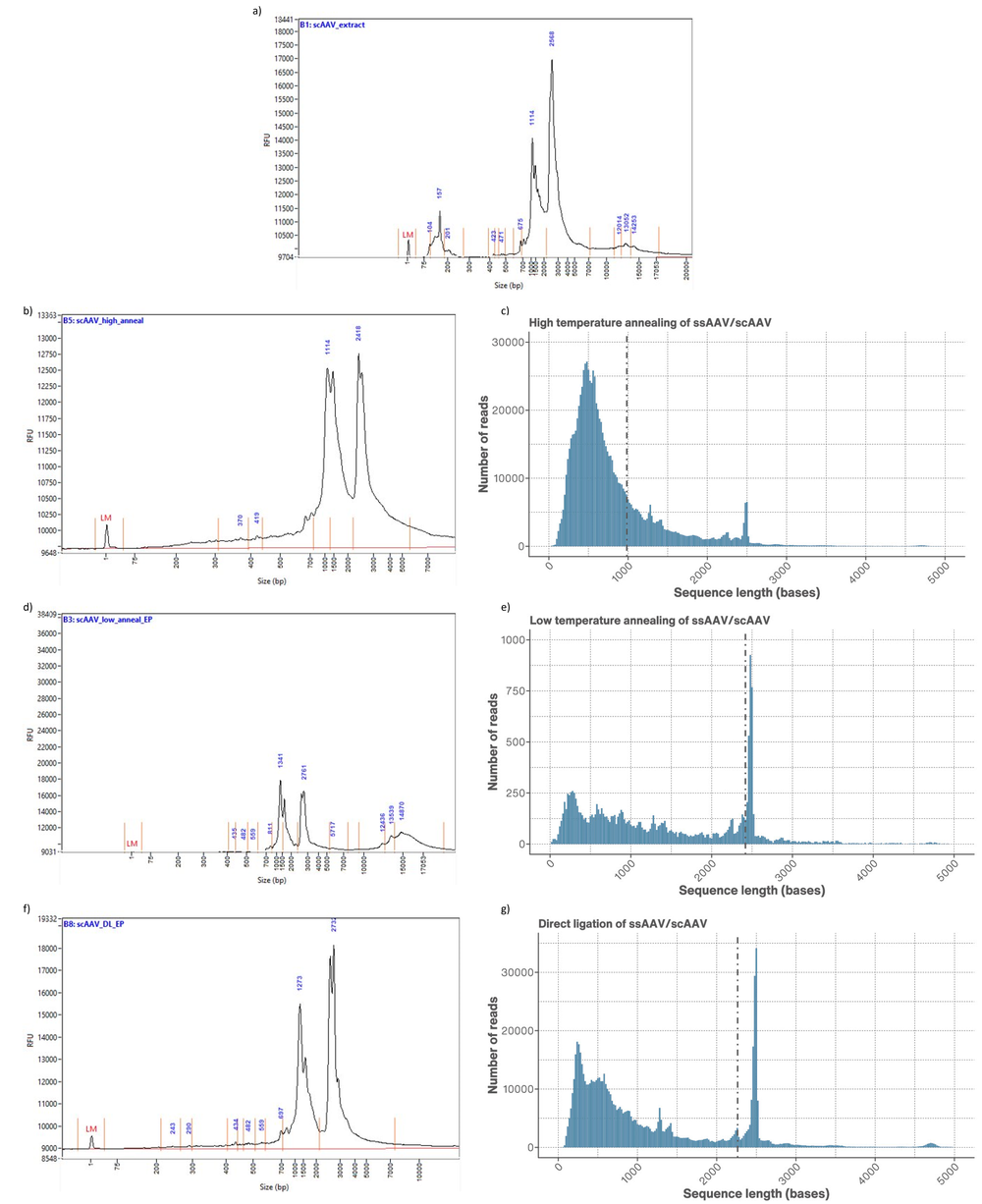 Figure 5. a) Femtopulse trace of scAAV present in the initial AAV extract trace. b) Femtopulse trace of ssAAV annealed using the high temperature method compared to c) the read length distribution of the same ssAAV/scAAV sample. d) Femtopulse trace of ssAAV annealed using the low temperature method compared to e) the read length distribution of the same ssAAV/scAAV sample. f) Femtopulse trace of ssAAV annealed using direct ligation compared to g) the read length distribution of the same ssAAV/scAAV sample.
Figure 5. a) Femtopulse trace of scAAV present in the initial AAV extract trace. b) Femtopulse trace of ssAAV annealed using the high temperature method compared to c) the read length distribution of the same ssAAV/scAAV sample. d) Femtopulse trace of ssAAV annealed using the low temperature method compared to e) the read length distribution of the same ssAAV/scAAV sample. f) Femtopulse trace of ssAAV annealed using direct ligation compared to g) the read length distribution of the same ssAAV/scAAV sample.
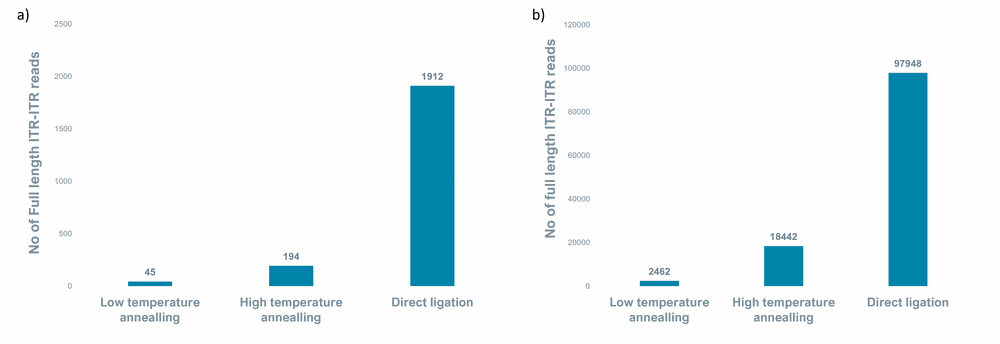 Figure 6. Number of full-length ITR-ITR reads in a) ssAAV and b) ssAAV and scAAV samples for all annealing methods.
Figure 6. Number of full-length ITR-ITR reads in a) ssAAV and b) ssAAV and scAAV samples for all annealing methods.
Rapid barcoding DNA V14 (SQK-RBK114.24 or SQK-RBK114.96) library preparation
For plasmid sequencing, we recommend using the Rapid Barcoding Kit 24 V14 (SQK-RBK114.24). Using the same sequencing kit, we investigated if we could effectively sequence full length AAV genomes. The results showed significantly fewer full-length AAV genomes compared to sequencing with the Native Barcoding Kit 24 V14 (SQK-NBD114.24). We also noted the identification of alternative AAV genome configurations in both preparations, confirming these genomes are present in AAV extracts and are not an artefact from the library prep. Both library preps were sequenced with HAC basecalling for ~40 hours.
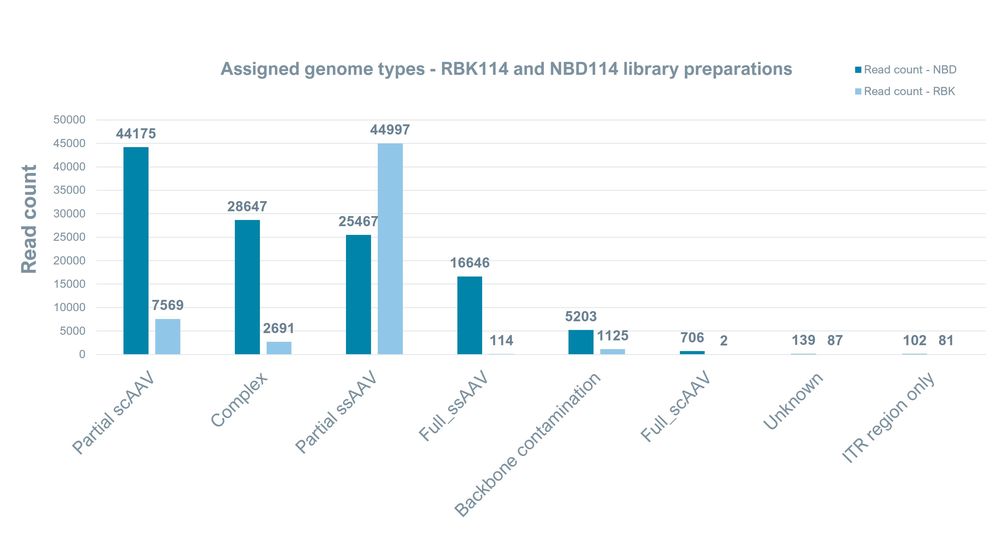 Figure 7. Read counts for identified AAV structures present in the AAV libraries prepared with SQK-NBD114.24 and SQK-RBK114.24 sequencing kits.
Figure 7. Read counts for identified AAV structures present in the AAV libraries prepared with SQK-NBD114.24 and SQK-RBK114.24 sequencing kits.
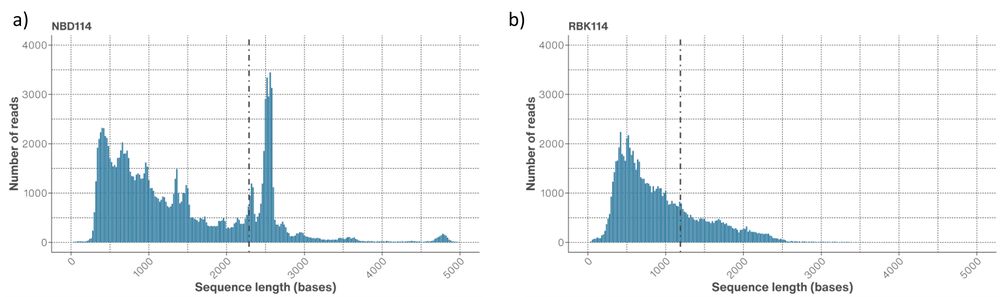 Figure 8. Read length distributions of the AAV extracts prepared with a) SQK-NBD114.24 and b) SQK-RBK114.24 sequencing kits. The dotted line represents the N50 for all reads.
Figure 8. Read length distributions of the AAV extracts prepared with a) SQK-NBD114.24 and b) SQK-RBK114.24 sequencing kits. The dotted line represents the N50 for all reads.
Notes on downstream analysis of AAV
Backbone contamination This is when part of the transgene plasmid backbone (i.e. outside of the ITR-ITR region) is incorporated in an alignment. This may be either a few bases or a large quantity of the DNA.
If a high level of backbone contamination is observed, we recommend checking the starting position of ITR1 and the end position of ITR2 to ensure it is base accurate. Additionally, IGV (Integrative Genomics Viewer) can be used to see how far beyond the read end positions are in the ITR regions and their consistency.
Defining full-length ssAAV in EPI2ME There is a user-defined threshold (itr_fl_threshold) that allows you to define how many bases from the outer end of each ITR the EPI2ME workflow can miss while still defining a read as full-length. The default is 100 bases from the outer end of both ITRs. The smaller the threshold, the stricter the full-length condition is.
Full-length ITR-ITR reads are calculated using passed reads that span the reference with a maximum of 100 bases missing from the outer end of each ITR. As mentioned above, this parameter (itr_fl_threshold) can be defined to modify the number of bases missing from an ITR, ensuring that the region between the two ITRs is classified as complete.
Summary
To maximise the number of full-length AAV genomes for sequencing to generate high quality data for analysis, we recommend using the PureLink™ Viral RNA/DNA Mini Kit alongside DNaseI treatment and skipping the self-annealing steps. These recommendations have been incorporated into our Adeno-associated virus (AAV) sequencing from recombinant adeno-associated virus (rAAV) vectors using SQK-NBD114.24 protocol.






