Blog: Plant genome hybrid assembly with MaSuRCA
In the first of a new series of blogs from members of the Nanopore Community, Aleksey Zimin from Johns Hopkins University discusses his software MaSuRCA, which uses long nanopore reads to enhance the contiguity of plant genomes.
 Aleksey ZiminI have been working in the field of Bioinformatics since 2002, beginning with my collaborations with The Institute for Genomic Research (TIGR) and Celera Genomics. The main goals of my research are (i) developing algorithms and software for de novo genome assembly for the latest generation sequencing data and (ii) applying the software to produce de novo assemblies for the most challenging genomes. I lead the development of the MaSuRCA genome assembly package, which is currently able to produce accurate high-quality assemblies from sequencing data produced by long- and short-read technologies. MaSuRCA is free, open-source software available from GitHub at http://github.com/alekseyzimin. I maintain a blog dedicated to assembler development and results at http://masurca.blogspot.com. I have played a leading role in producing assemblies for many challenging genome projects, including the 26 Gbp genome of coast redwood (Sequoia sempervirens), the 17 Gbp genome of bread wheat (Triticum aestivum), the 3 Gbp Atlantic salmon (Salmo salar), and many other plants and animals.
Aleksey ZiminI have been working in the field of Bioinformatics since 2002, beginning with my collaborations with The Institute for Genomic Research (TIGR) and Celera Genomics. The main goals of my research are (i) developing algorithms and software for de novo genome assembly for the latest generation sequencing data and (ii) applying the software to produce de novo assemblies for the most challenging genomes. I lead the development of the MaSuRCA genome assembly package, which is currently able to produce accurate high-quality assemblies from sequencing data produced by long- and short-read technologies. MaSuRCA is free, open-source software available from GitHub at http://github.com/alekseyzimin. I maintain a blog dedicated to assembler development and results at http://masurca.blogspot.com. I have played a leading role in producing assemblies for many challenging genome projects, including the 26 Gbp genome of coast redwood (Sequoia sempervirens), the 17 Gbp genome of bread wheat (Triticum aestivum), the 3 Gbp Atlantic salmon (Salmo salar), and many other plants and animals. Plant genomes still present a significant challenge for assembly projects. They vary greatly in size, from hundreds of megabases to tens of billions of bases, and often contain rich repetitive sequence families. Furthermore, for many non-model plants, it is not practical to inbreed them due to their long reproductive cycle and thus an assembly algorithm has to deal with heterozygosity in the genome, where the maternal and paternal chromosomes differ in many locations, thus effectively increasing the “unique” genome size.
Due to high repeat content and heterozygosity, assemblies of plant genomes that use only data from a short-read technology are typically very fragmented, which presents challenges for downstream analyses, e.g. frequently exons belonging to the same gene end up in different scaffolds, and this sometimes results in these exons being annotated as two or more different genes. Long-read sequencing technology developed by Oxford Nanopore Technologies allowed for a breakthrough in plant genome assembly due to long read lengths. Long reads span repeats, thus allowing for correct assemblies of complex repetitive regions, because the only way to assemble a repeat reliably is to have a read that is longer than the repetitive element, thus allowing to compute overlaps between the reads by using anchors from the unique sequence around the repeat.
Long reads are still a lot more expensive than short reads per base, and they come with an additional challenge of higher error rates. This raises the coverage requirements for assembly projects because short overlaps between these reads cannot be detected reliably, and thus any assembly using only long reads has to have at least 50x coverage of the genome, which puts strenuous requirements on the hardware and algorithms for big plant genomes. In many cases, short reads are still needed for post-assembly “polishing”, where the short reads are aligned to the assembled consensus in order to correct the errors that remain.

The hybrid assembly approach provides a way to simplify and reduce the cost of the assembly by reducing long-read coverage requirements. This approach first uses short reads to correct sequencing errors in nanopore data and then creates an assembly from the corrected reads. Such a technique is implemented in the publicly available, open-source MaSuRCA (Maryland Super-Read CAbog) assembler available on GitHub: https://github.com/alekseyzimin/masurca (Zimin et al., 2017). MaSuRCA requires 60-100x genome coverage from 150 bp paired-end short reads in addition to >10x coverage from nanopore reads to produce a contiguous assembly of a genome. The core idea in MaSuRCA is the super-reads technique, where k-mers (all sub-sequences of length k) from short reads are used to extend each short read uniquely in both forward and reverse directions. The extended reads are called super-reads. Typically, many short reads extend to the same super-read, and thus we replace them all by the corresponding super-read. The super-reads are much longer, averaging 400-2000 bases depending on the genome, and there are much fewer of them – they typically cover the genome at 2-4x. In the Loblolly pine project (Zimin et al., 2014), we were able to transform 15 billion short reads into 150 million super-reads. The super-reads are much better suited for correcting the long reads due to their longer lengths and lower coverage. MaSuRCA then uses each long read as a template by computing the approximate alignments of all super-reads to the long read and then looking for the best path of the exactly overlapping aligned super-reads that spans the long read. Such a path is called a “mega-read”. These mega-reads typically have a very low error rate, less than 1%, and most of them span the full length of the long read. Then, the mega-reads are assembled in MaSuRCA by either a customised version of the CABOG assembler (Miller et al., 2008), or the Flye assembler (Kolmogorov et al., 2019). Both assemblers are supplied and installed with MaSuRCA, so the user does not need to deal with external dependencies. The MaSuRCA mega-reads algorithm is illustrated in Figure 1.
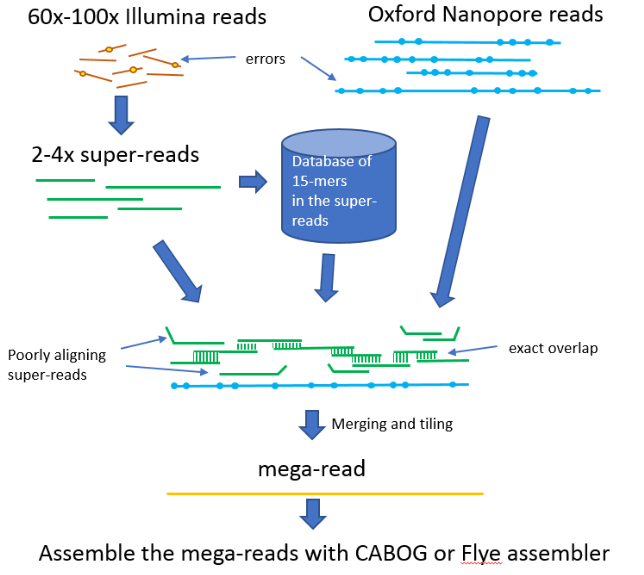
| Giant sequoia assembly | |||||
|---|---|---|---|---|---|
| Sequence in contigs (Gbp) | N50 contig (Kbp) | N50 Scaffold (Kbp) | Number of contigs | Number of scaffolds | |
| Short read only | 7.9 | 12 | 65 | 2,507,175 | 1,007,217 |
| Short read + Nanopore long read | 8.1 | 360 | 489 | 49,676 | 39,821 |
| Coast redwood assembly | |||||
| Short read only | 24.3 | 1.2 | 2.3 | 36,917,629 | 27,303,833 |
| Short read + Nanopore long read | 26.5 | 97 | 110 | 549,274 | 518,205 |
Table 1. Giant Sequoia and coast redwood assemblies. Using Oxford Nanopore long reads results in much more complete and contiguous assembly compared to short read only.
The MaSuRCA algorithm is designed to be able to perform a 30 Gbp plant genome assembly on one computer with 2 TB of RAM. A cluster can be used as well, but the main execution node must meet the RAM requirements. The memory requirement is approximately linear in genome size, and we were able to assemble the 16 Gbp genome of bread wheat on a cluster of computers with maximum memory usage of 1 TB. For a successful genome project, I recommend starting with 60x to 100x coverage with 150 bp or longer paired-end short reads (400-500 bp fragment size recommended), and at least 20x coverage by Oxford Nanopore reads. We used this sequencing strategy followed by assembly with MaSuRCA in the Redwood genome project funded by Save the Redwoods League and headed by David Neale and Steven Salzberg, resulting in high-quality assemblies of ~8 Gbp giant sequoia and ~26 Gbp coast redwood genomes. In Table 1, I compared the short read + long read assemblies to the ones produced using short-read data alone. The assemblies that use long reads are 30+ times more contiguous and more complete.
I am actively developing and maintaining MaSuRCA. In the past year I managed to increase the assembly speed by a factor of two, while at the same time improving the contiguity of the resulting assemblies. I am currently adding additional post-processing tools such as a chromosome scaffolder script that allows using a closely related genome sequence that is placed on the chromosomes to scaffold the assembled contigs into chromosome-sized scaffolds, a very efficient polishing tool called POLCA (Zimin and Salzberg, 2019).
References
- Zimin AV, Puiu D, Luo MC, Zhu T, Koren S, Marçais G, Yorke JA, Dvořák J, Salzberg SL. Hybrid assembly of the large and highly repetitive genome of Aegilops tauschii, a progenitor of bread wheat, with the MaSuRCA mega-reads algorithm. Genome Research. 2017 May 1;27(5):787-92
- Zimin A, Stevens KA, Crepeau MW, Holtz-Morris A, Koriabine M, Marçais G, Puiu D, Roberts M, Wegrzyn JL, de Jong PJ, Neale DB. Sequencing and assembly of the 22-Gb loblolly pine genome. Genetics. 2014 Mar 1;196(3):875-90.
- Kolmogorov M, Yuan J, Lin Y, Pevzner PA. Assembly of long, error-prone reads using repeat graphs. Nature biotechnology. 2019 May;37(5):540.
- Miller JR, Delcher AL, Koren S, Venter E, Walenz BP, Brownley A, Johnson J, Li K, Mobarry C, Sutton G. Aggressive assembly of pyrosequencing reads with mates. Bioinformatics. 2008 Oct 24;24(24):2818-24.
- Zimin AV, Salzberg SL. The genome polishing tool POLCA makes fast and accurate corrections in genome assemblies. bioRxiv. 2019 Jan 1.






