Nanopore Community Meeting 2021: Day 3
With an incredible attendance and incredible content on Day 2 of the Nanopore Community Meeting 2021, Day 3 saw a change in tracks: Human & translational research remained along with Plant & Animal; Bioinformatics took over from Microbiology & Metagenomics.
Spotlight session winner - Phages‚ faeces‚ and PromethION: using nanopore to investigate the cattle slurry virome - Ryan Cook (University of Nottingham)
Shortly after the start of the day we heard from our Spotlight session winner, Ryan Cook. After his pitch on day 2, the NCM 2021 delegates had decided they wanted to hear from Ryan the most, and he took to the cross-track stage to deliver his talk on the use of PromethION for investigating the cattle slurry virome.
As most people are aware of, cows produce enormous amounts of waste. This takes the form of faeces, urine, and as a result also bedding. All of this is is collected into a large slurry tank, with the high phosphate and nitrate content of the waste meaning it can be used as an agricultural fertilizer. This makes the slurry incredible useful in its applications, but 'we know incredibly little about its microbial composition’.
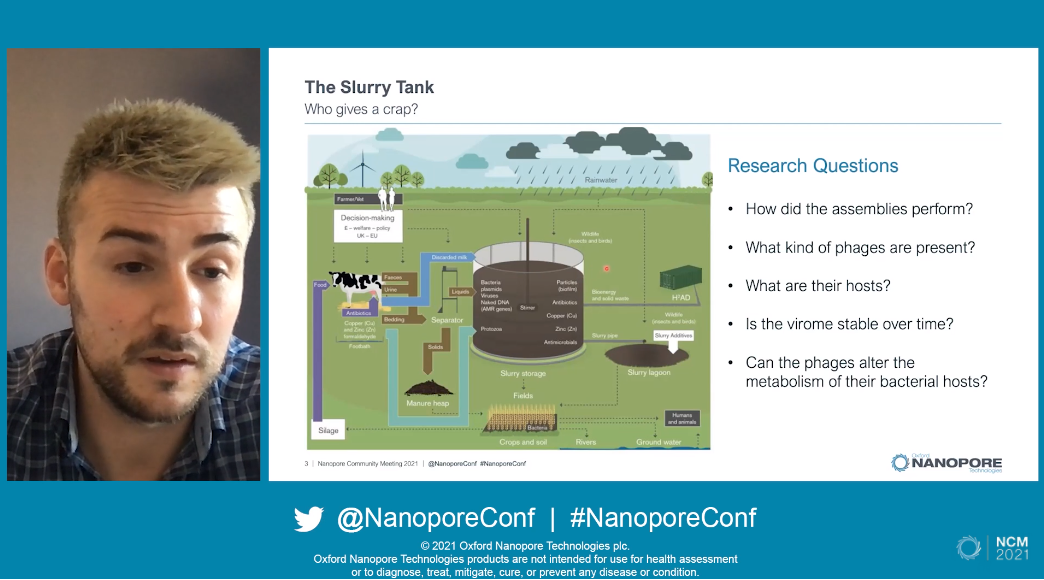 Ryan detailed how he obtained slurry tank samples and sequenced the viral fraction using short-read and long-read sequencing. Five viral DNA samples underwent short-read sequencing, as well as being pooled, amplified, and sequenced using PromethION. With the resultant data he carried out viral genome assmebly, and found assemblies generated with long-read nanopore seuqencing were approximately 2kb longer than their short-read counterparts. Looking at the protein expression characteristics, the average open reading frame predicted using PromethION technology was 85 amino acids, which further increased to 103 aa if polished with short-read sequences. Investigations uncovered this was due to the removal of erroneous stop codons, which in turn helped with protein prediction and functional annotation of the assemblies.
Ryan detailed how he obtained slurry tank samples and sequenced the viral fraction using short-read and long-read sequencing. Five viral DNA samples underwent short-read sequencing, as well as being pooled, amplified, and sequenced using PromethION. With the resultant data he carried out viral genome assmebly, and found assemblies generated with long-read nanopore seuqencing were approximately 2kb longer than their short-read counterparts. Looking at the protein expression characteristics, the average open reading frame predicted using PromethION technology was 85 amino acids, which further increased to 103 aa if polished with short-read sequences. Investigations uncovered this was due to the removal of erroneous stop codons, which in turn helped with protein prediction and functional annotation of the assemblies.
Turning the comparison to ‘read recruitment’, the results suggested that long-read sequencing was far better than short-read alone and could be further improved with polishing and de-replicating hybrid contigs. When read recruitment was normalised with phage abundance, it became possible to predict the diversity within the slurry tank. The long-read contigs estimated far higher biodiversity than short-read, suggesting that just using short-read data could lead to underestimation of diversity within an environment. Ryan also estimated how complete the phage genomes were by comparing the short-read sequences and the polished PromethION results. A higher proportion of medium- and high- quality genomes were detected in the assemblies containing the long reads (and a lower proportion of low-quality) compared with short-read data only. The slurry tank contained 7,682 viruses, and ‘98% of the tank was dominated by novel genera’. A number of interesting features which could alter the metabolism of the host were identified, including virulence factors and anti-microbial resistance genes.
Panel Plenary - Pushing the limits of clinical research
From an early-career researcher, we segued into a panel plenary discussion with some of the most impressive characters using nanopore sequencing in the translational human research field. Areeba Patel, Thidathip Wongsurawat, Franz-Josef Müller, Helene Kretzmer and Luna Djirackor held a fascinating discussion on how to push the boundaries of the use of using nanopore sequencing in clinical research, but prior to this we heard their talks. All were on the topic of classifying central nervous system (CNS) tumours using nanopore sequencing technology.
Rapid-CNS²: rapid‚ comprehensive adaptive nanopore sequencing of CNS tumors — a proof-of-concept study - Areeba Patel (German Cancer Research Center (DKFZ) &
University Hospital Heidelberg)
 Areeba opened by stating that CNS tumours are some of the hardest to treat. They are also some of the hardest to classify: this is evidenced by the current WHO 2021 classification incorporating mutations, copy number alterations, and target gene methylation status into its criteria. With this many considerations it can take around 22 days to classify a tumour, including time taken to even obtain the necessary number of samples. Areeba's long-term aim therefore is to develop a classification solution which is accessible and affordable, steps for which she has already begun with her use of the ReadFish algorithm for adaptive sampling, targeting a neuropathology gene panel and CpG sites relevant for methylation classification. She has termed this workflow Rapid-CNS2, available on Github now, and consisting of 2 parallel analysis workflows: 1) methylation calling withh Megalodon, 2) variant calling including SNV, copy number variant, and structural variant calling.
Areeba opened by stating that CNS tumours are some of the hardest to treat. They are also some of the hardest to classify: this is evidenced by the current WHO 2021 classification incorporating mutations, copy number alterations, and target gene methylation status into its criteria. With this many considerations it can take around 22 days to classify a tumour, including time taken to even obtain the necessary number of samples. Areeba's long-term aim therefore is to develop a classification solution which is accessible and affordable, steps for which she has already begun with her use of the ReadFish algorithm for adaptive sampling, targeting a neuropathology gene panel and CpG sites relevant for methylation classification. She has termed this workflow Rapid-CNS2, available on Github now, and consisting of 2 parallel analysis workflows: 1) methylation calling withh Megalodon, 2) variant calling including SNV, copy number variant, and structural variant calling.
The pipeline's performance on 35 glioma research samples showed higher resolution than the standard panel sequencing methods, and were comparible to the gold standard EPIC arrays. Overall, complete concordance of results from Rapid-CNS2 compared to convential methods was seen in the majority of research samples analysed.This all came with the beenfits of low capital cost, in one assay combining several results, and delivering drastically reduced turnaround times.
Nanopore-based copy number variation approach for adult glioma classification — WHO 2021 - Thidathip Wongsurawat (Mahidol University)
 Following Areeba, Thidathip introduced her research into the potential for nanopore sequencing to perform copy number variation-based calssification of adult glioma, in line with WHO 2021 criteria. In 2007 the WHO glioma classification criteria were based entirely on phenotype; in 2016 genomic data was incoroprated by the WHO for the first time; and finally in 2021 the guidelines included an extended list of genetic markers. A new marker, a CDKN2A/B deletion, has just been added but has no test for its identification currently. More concerningly FISH is commonly used for tumour classification but there is no standard method for identifying the CDKN2A/B deletion detection with FISH. Thidathip wants to develop an approach for just this, but crucially one that is simple to perform, has a quick turnaround time and, ideally, is more informative than FISH. She believes nanopore sequencing could provide this.
Following Areeba, Thidathip introduced her research into the potential for nanopore sequencing to perform copy number variation-based calssification of adult glioma, in line with WHO 2021 criteria. In 2007 the WHO glioma classification criteria were based entirely on phenotype; in 2016 genomic data was incoroprated by the WHO for the first time; and finally in 2021 the guidelines included an extended list of genetic markers. A new marker, a CDKN2A/B deletion, has just been added but has no test for its identification currently. More concerningly FISH is commonly used for tumour classification but there is no standard method for identifying the CDKN2A/B deletion detection with FISH. Thidathip wants to develop an approach for just this, but crucially one that is simple to perform, has a quick turnaround time and, ideally, is more informative than FISH. She believes nanopore sequencing could provide this.
For her research she had access to 19 samples known to have the IDH mutations, which are included in the current typical testing algorithm for adult glioma. By adapting SMURF-seq, and using the MinION devices followed by CNV calling, she detected 1p deletion, 19q deletion, and CDKN2A/B deletion. This answer could be obtained after even just 8 minutes of sequencing (it can take longer to drink a cup of tea!). After analysis with all 19 samples, her nanopore-based method showed 100% concordance with current testing methods, but crucially at a lower cost. Thidathip took great delight in pointing out also that, thanks to its use of MinION, almost any lab world-wide can access this (and they also got the same results with Flongle). In a hypothetical workday, her process could start at 9 am and you could have your CNV profile by 1pm - even including an hour for lunch.
Real-time cancer classification with nanopore sequencing - Franz-Josef Müller (University Hospital Schleswig-Holstein) & Helene Kretzmer (Max Planck Institute for Molecular Genetics)
 Doubling the numbers of our speakers in one swoop, next we heard from Franz-Josef and Helene on their work for real-time cancer classification. In fact rather than just heard from, we saw from, as the first half of their shared talk focused on an engaging video demonstration of their lab workflow for brain tumour classification based on a sample's methylation profile.
Doubling the numbers of our speakers in one swoop, next we heard from Franz-Josef and Helene on their work for real-time cancer classification. In fact rather than just heard from, we saw from, as the first half of their shared talk focused on an engaging video demonstration of their lab workflow for brain tumour classification based on a sample's methylation profile.
Franz-Josef spoke first, introducing their setup which had been optimised for the fastest possible classificaton of brain tumour types: they prepare libraries with the rapid barcoding kit before sequencing on the MinION Mk1B and performing analysis with Megalodon. He then moved smoothly into their 'Clinical Demonstrator' video, so called as at the core of their work lies the desire to transition their research on epigenetic features of cancer subtypes into the clinic. Biopsies fresh from the theatre were bought to the sequencing lab, prepared for sequencing, and then all analysis was carried out locally. 44 minutes after biopsy processing started, they see their first prediction on classification type come in.
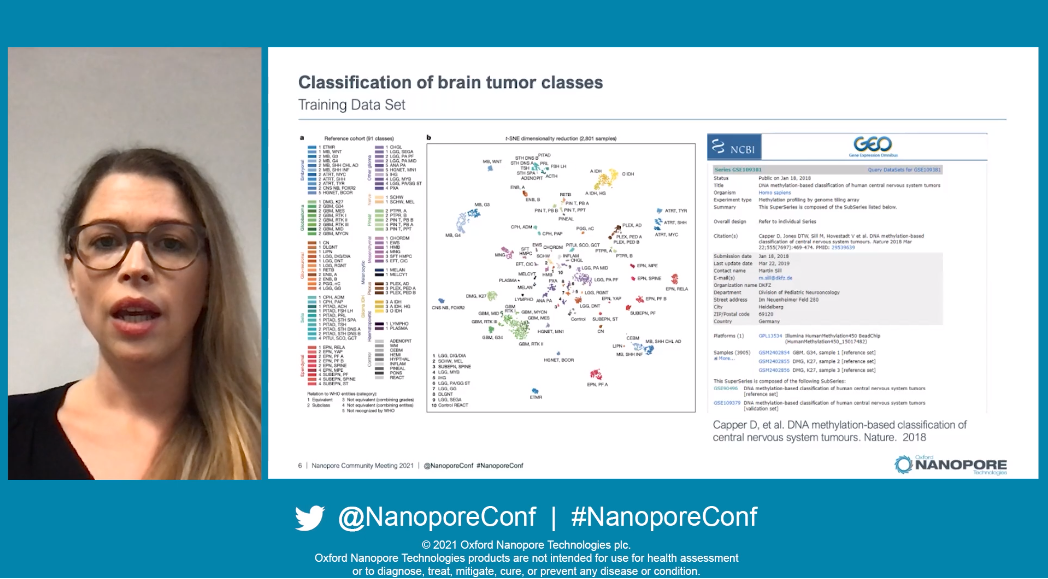 Helene then took over to explain that their CpGs are filtered within their pipeline to those covered by commercially available methylation arrays. In real-time, they see ~500 CpG sites sequenced within the first few minutes of a MinION run, rising to 2,000-5,000 within the first 30 minutes. At 4-5 hours, no new CpGs are sequenced. Building on research by their peers, their training set for classification consists of >2,800 samples and covers over 90 different tumour classes - very comprehensive.
Helene then took over to explain that their CpGs are filtered within their pipeline to those covered by commercially available methylation arrays. In real-time, they see ~500 CpG sites sequenced within the first few minutes of a MinION run, rising to 2,000-5,000 within the first 30 minutes. At 4-5 hours, no new CpGs are sequenced. Building on research by their peers, their training set for classification consists of >2,800 samples and covers over 90 different tumour classes - very comprehensive.
Helene concluded by sharing some example result, stating that they see a stable classification of brain tumour class after only 1 hour of sequencing, and showing the read out from their pipeline includes a list of tumour classes, as well as the classificaiton probabilities.
Discussion - Joined by Luna Djirackor (Oslo University Hospital)
After three thoroughly impressive talks demonstrating the innovative research this group are carrying out, they were joined by an incredibly worthy peer in the form of Luna Djirackor, who at London Calling 2021 presented her work on the potential of intraoperative (yes, whilst they are on the operating table!) nanopore-based methylation classification of brain tumours.
 A question first on library prep kits and workflows, Areeba noted that one of the reasons adaptive sampling had been so attractive to her was, well - there's no different sample preparation methods required! The amount of data they required - their coverage - was up next, with different answers for different applications. Areeba had sufficient data for variant calling at 10x, but could conduct methylation profiling with just one read. One. Read. Thidathip required less than 1x depth of coverage to obtain the correct result, while Luna stated that to get results back during surgery they needed ~ 3,500 CpG sites called. Helene determines CpG methylation as a binary result, and single sites were rarely covered more than once during their sequencing run but this was sufficient for classification.
A question first on library prep kits and workflows, Areeba noted that one of the reasons adaptive sampling had been so attractive to her was, well - there's no different sample preparation methods required! The amount of data they required - their coverage - was up next, with different answers for different applications. Areeba had sufficient data for variant calling at 10x, but could conduct methylation profiling with just one read. One. Read. Thidathip required less than 1x depth of coverage to obtain the correct result, while Luna stated that to get results back during surgery they needed ~ 3,500 CpG sites called. Helene determines CpG methylation as a binary result, and single sites were rarely covered more than once during their sequencing run but this was sufficient for classification.
Moving onto data analysis and infrastructure, for Franzef and Helene everything was carried out locally. Areeba used a cluster and a local workstation, with the main compute requirement for adaptive sampling being a GPU. Thidathip turned to a bioinformatician for analysis, but hopes to use real-time analysis in the future. Luna echoed Franzef and Helene, with a local setup including a GPU laptop, a MinIT, and a MinION.
Luna, Franzef and Thidathip all used a MinION in their research, but saw the benefits in GridION for allowing multiple on-demand experiments to take place. Areeba agreed that the flexibility of the GridION was key. Further savings in time and cost could be achieved with multiplexing on a single flow cell, or washing a flow cell and re-using for later libraries.
The last topic of discussion was whether any training would potentially be needed for the real-world implementation of their workflows. Franzef emphasized the need for good and reliable ways to communicate certainty, as well as uncertainty, of the results. Areeba explained how the report outputted from her team’s workflow is as comprehensive as possible, detailing not just what tumour class has been detected, but also what might those results imply, and the level of certainty.
The annotation of novel genes in a complete human genome - Alaina Shumate (Johns Hopkins University, USA)
 The conclusion of Alaina's talk was pleasingly succinct: Quality annotation of genomes is dependent on quality assemblies, and this has been made possible with the advent of long-read sequencing. But what was the content which lead to this summation?
The conclusion of Alaina's talk was pleasingly succinct: Quality annotation of genomes is dependent on quality assemblies, and this has been made possible with the advent of long-read sequencing. But what was the content which lead to this summation?
As most readers will be aware, a recent telomere to telomere (T2T) assembly of the human genome has been completed. Ultra-long nanopore reads were instrumental in this, and the group's efforts yielded a particularly impressive maximum read length of 1.3 Mbp (a true blue whale). As part of this, a number of novel sequence features were identified, including 1,956 novel genes within which were 99 protein-coding genes exclusively found in the T2T assembly. Finding these novel genes was the focus of Alaina's talk.
Alaina saw the need for a tool that could map, or lift over, gene annotations from a reference genome to an improved assembly of the same (or a closely related) species. For this, she has been developing a tool, Liftoff, with a key feature of being able to use reference annotations to find additional paralogs in a new assembly. It was designed to address some of the issues with existing lift over tools, which convert single coordinates that are not sufficient when, with gene annotations, you are mapping genomic intervals.
Numerous challenges exist during lift over processes, including when genes align in fragments or when a gene has more than one alignment but none of them, at first glance, appear to be the most appropriate to settle upon. Logic that aids in resolving this involves ignoring intron alignments at later stages of analysis: they are important in the initial alignment step, but are not useful for coordinate conversion. With these approaches, it is easier to determine which combination of alignments contain all of the exons in the correct order. Other factors are incorporated into the Liftoff tool, including a tunable parameter to avoid the generation of transcripts that aren't biologically feasible due to the distance from start to end.
With the use of this tool, paralogs can also be identified. Following mapping of reference annotations onto the new genome, they go back to minimap2 to look for additional alignments. Alignments over already annotated genes are removed, then the lift over process is repeated and paralogs are labelled if above a sequence identity threshold of 95%. One limitation of liftoff here is that it relies on a reference annotation and can only identify new genes that are paralogs of those in the reference. Using this approach and the 5,175 medically relevant genes compiled by the Genome in a Bottle Consortium, Alaina identified clinically relevant paralogs from the T2T assembly including the potent ligand for CCR5 HIV-1 coreceptor, CCL3L1 (a copy number of which is correlated to a lower susceptibility to HIV-1). And with that, Alaina reached her conclusion.
Affordable GPU compute makes nanopore sequencing even more disruptive and empowering - Miles Benton (Institute of Environmental Science and Research)
 Following on from Alaina, Miles Benton took us into the world of more consumer-based technology, and his adventures and developments in using GPU compute to make nanopore sequencing even more empowering than it's already believed to be.
Following on from Alaina, Miles Benton took us into the world of more consumer-based technology, and his adventures and developments in using GPU compute to make nanopore sequencing even more empowering than it's already believed to be.
Miles' inspiration for his work is close to his heart, as it is for those who hear his story. His son was born premature and suffering from suspected meningitis, and confirmation of this required a lengthy wait for a PCR result. Miles' questioned this necessity, and with his knowledge of nanopore sequencing, he wondered why we couldn't see a future with this amazing technology available at the bedside. If he could combine nanopore sequencing with an affordable GPU computer then he could enable robust, accessible, real-time sequencing pipelines at low cost. Maybe in the future, another parent will not have to wait as he did.
Taking inspiration from the inclusion of on-board GPUs in many of the sequencers provided by Oxford Nanopore, Miles investigated the hardware options available in the consumer electronics market and believes he can create a viable, highly accessible setup for real-time basecalling of a MinION for $1,100. He began to test this out and hit some initial hurdles with software integration. As is often seen however the nanopore community on Twitter helped to chip in with piecing together a solution, and he was able to generate a robust enough device that, not only can it basecall data, it was also running his presentation to the Nanopore Community meeting at the same time.
Proof of principle achieved, Miles took a look at the wider picture. And the picture looked back. Lim Fang-Shiang, a Ph.D student from Germany, reached out to Miles seeking his advice for combining a Jetson AGX unit to a MinION. Shortly after this, collaborators sought the help of Lim to identify a pathogen that was killing off insects in their colony and within 24 hours that had a pathogen ID, with a fully assembled genome, at the site of the problem. Miles felt the buzz of this involvement.
Devin Drown was another purveyor of Miles' skills, as he compiled a solution for Devin's high school project with the MinION and the Jetson NX to help empower and excite the next generation of students about what is possible. Adaptive sampling was even successfully used, a topic on the periphery of Miles' talk but still something he finds amazing.
Miles is not alone however, and he exalted the work of another instrumental figure in pairing the MinION with the Nvidia Jetson offering: Jurgen Hench, from the University of Basel, who adapted the technology for the work introduced by Luna Djirackor to classify brain tumours intraoperatively. Bringing it back into the clinical research setting, Miles now considers what he could further create that would help to identify what was in the cerebrospinal fluid of his son. His enthusiasm and passion is infections and after just one meeting with Miles, a local pathologist purchased a MinION and a Jetson AGX. Together, they envisage a future where real-time diagnosis may be possible.
Towards comprehensive genetic diagnosis of repeat expansion disorders with targeted nanopore sequencing – Ira Deveson (The Garvan Institute of Medical Research)
 On the home straight of our talks at NCM 2021 now, we moved away from the Bioinformatics track to our final cross-track plenaries. Ira Deveson, the leader of the Genomic Technologies research group, is currently focusing on applying and developing long-read sequencing technologies in a diverse set of research areas. And what better place to start, than with adaptive sampling.
On the home straight of our talks at NCM 2021 now, we moved away from the Bioinformatics track to our final cross-track plenaries. Ira Deveson, the leader of the Genomic Technologies research group, is currently focusing on applying and developing long-read sequencing technologies in a diverse set of research areas. And what better place to start, than with adaptive sampling.
Requiring no special library preparation, adaptive sampling allows you to selectively target DNA sequences on-flow cell, leading to enrichment of what you want to see and depletion of what you don't, all through software. With this technique in hand, he and his group set about testing its potential for genotyping of short tandem repeat (STR) expansions - these are ~2-12 bp sequence motifs that are repeated multiple times in succession, making up ~7% of the human genome, which vary wildly in length between individuals. The variation in length can be healthy, but some expanded repeats can also act as pathogenic mutations. Many of the diseases caused by this are neurological and include Hungtington's Disease, Fragile X Syndrome, and ALS. At least 37 different genes with problematic repeat expansions are currently known to cause >40 neurological disorders.
Clinical diagnosis of repeat expansion disorders currently relies on labour-intensive techniques such as Southern blots, with each assay testing for only a single gene. Given the number of disorders associated with repeat expansions across many genes, which present with overlapping symptoms, clinicians currently have to make their best guess at the correct test to order which can result in a long wait for a diagnosis. Nanopore sequencing could in the future solve this problem in one step, by instead reading STRs end-end in single long reads, whilst adaptive sampling could negate the need to perform more costly whole-genome sequencing. So they decided to test this out, in conjunction with adaptive sampling, for parallel testing of all disease-associated STR genes in a single nanopore assay (this has flavours of the cancer gene panel approach taken by Phill James in the Nanopore Apps update: see Day 2 write up). Their chosen implementation of adaptive sampling was through ReadFish.
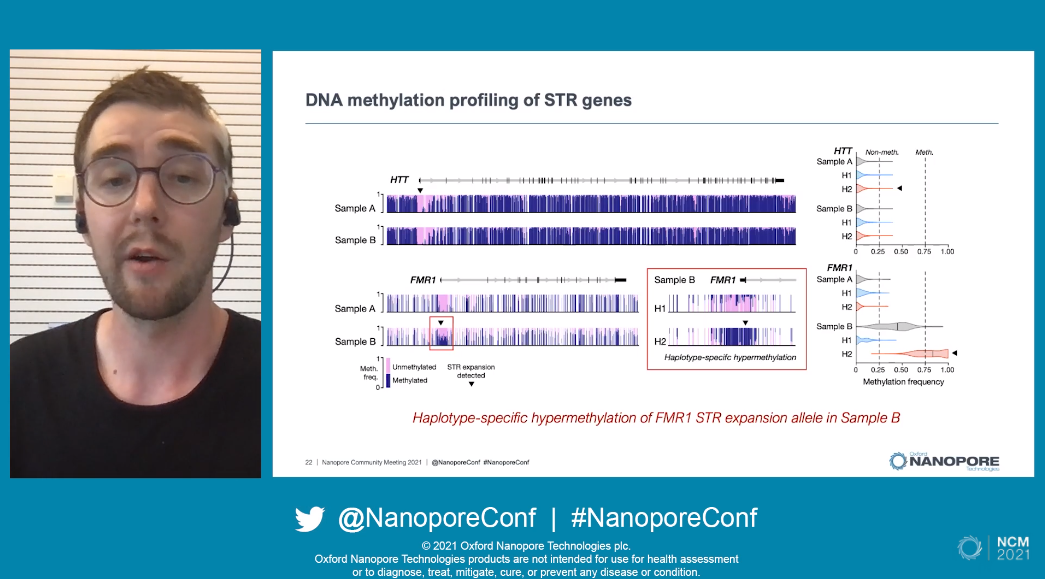
Their experiment began by cataloging all known neurological-disease-implicated STR genes and their flanking regions, which formed the targets for enrichment via adaptive sampling. An example of this being successfully implemented was shown by Ira from the HTT gene, displaying clear enrichment across the gene and lower read depth from the adjacent regions. They saw a depth of coverage of 25-35x for their targets from a single MinION Flow Cell - similar to that produced with whole-genome sequencing on a PromethION flow cell.
With the HTT gene as a focus, Ira and his team performed phasing and assembled the consensus repeat sequence for each parent. An example of this was given where one parent had a healthy copy with 18 CAG repeats, while the other had a pathogenic allele with 64 copies. In the 12 samples they tested, the adaptive sampling results correctly matched the healthy or diseased phenotypes from the clinical research samples. In fact, the STR copy numbers identified by the clinical tests were almost identical to those from nanopore sequencing, demonstrating that sequencing was at least as precise as the current, established methodologies. Some genes such as FMR1 and RFC1 cannot rely on repeat length for the identification of pathogenic variants however.
Turning attention to epigenetic modifications, these can conveniently be identified alongside nanopore sequencing. Methylated regions which are believed to be pathogenic were identified in the FMR1 region, as an example, which would lead to the silencing of this region and just so happens to be considered the main mechanism of pathogenicity in Fragile X syndrome. Ira stressed that characterizing this feature could not be done easily with any other technology. Similar real-world beneficial results were reported for the RFC1 gene, in which STR expansions are the leading cause of a debilitating ataxia, CANVAS, and nanopore sequencing was able to succinctly identify all 3 features of the pathogenic variant which currently require multiple molecular tests.
Further benefits of nanopore sequencing in these applications rely on the ability to assess several genes simultaneously, rather than one test at a time as is currently the way. This could allow for a greater understanding of the true genetic diversity at these repeat sites, which are currently poorly understood through the use of traditional sequencing technology. Where currently, the limited resolution of these genes means that the boundary between what is considered a pathogenic or non-pathogenic variant may not be clearly defined, Ira emphasized the future potential of adaptive sampling and nanopore sequencing to untangle this complex picture.
This brings us to the end of NCM 2021, we will be making content available on the Nanopore Resource Centre over the coming weeks, so please look out for talks as they become available.






