Blog: Copy number variation analysis from plasma: is it feasible using nanopore sequencing?
In this blog, Filippo Martignano shares his research into copy number variation (CNV) analysis in cancer, from cell-free DNA samples, using shallow whole-genome sequencing. Read on to hear why he chose nanopore technology for this project, and why his nanopore-based approach is ‘probably one of the fastest whole genome CNV analysis workflows’.
 Filippo Martignano I started my laboratory experience as a molecular biologist at the Istituto Scientifico Romagnolo per lo Studio e la Cura dei Tumori (IRST, Meldola, Italy), where I performed my internship for bachelor and masters degrees in Biotechnologies, before being hired as a research assistant. During the years spent at IRST I had the opportunity to gain expertise on several aspects of translational cancer research. I focused my research on the study of liquid biopsy biomarkers such as plasma/urinary microRNAs and cell-free DNA. In late 2017, I started my PhD programme in “Genetics, Oncology and Clinical Medicine” at the University of Siena (Italy) and Istituto per lo Studio la Prevenzione e la Rete Oncologica (ISPRO, Florence, Italy), under the guidance of Dr Silvestro Conticello (CNR-IFC). At ISPRO I had the opportunity to gain experience in next generation sequencing (short read and Nanopore), and computational approaches for the study of gene expression, DNA mutations, RNA editing, and copy number variation. This project is hence the result of the combination of all the skills I have gained so far, in both molecular and computational biology, with particular attention to eventual clinical applications.
Filippo Martignano I started my laboratory experience as a molecular biologist at the Istituto Scientifico Romagnolo per lo Studio e la Cura dei Tumori (IRST, Meldola, Italy), where I performed my internship for bachelor and masters degrees in Biotechnologies, before being hired as a research assistant. During the years spent at IRST I had the opportunity to gain expertise on several aspects of translational cancer research. I focused my research on the study of liquid biopsy biomarkers such as plasma/urinary microRNAs and cell-free DNA. In late 2017, I started my PhD programme in “Genetics, Oncology and Clinical Medicine” at the University of Siena (Italy) and Istituto per lo Studio la Prevenzione e la Rete Oncologica (ISPRO, Florence, Italy), under the guidance of Dr Silvestro Conticello (CNR-IFC). At ISPRO I had the opportunity to gain experience in next generation sequencing (short read and Nanopore), and computational approaches for the study of gene expression, DNA mutations, RNA editing, and copy number variation. This project is hence the result of the combination of all the skills I have gained so far, in both molecular and computational biology, with particular attention to eventual clinical applications. Why liquid biopsy?
In the “precision oncology” era, the characterisation of tumour genetic features is a pivotal step in cancer patients’ management. Typically, biopsy samples or surgical resections are necessary for biomarker investigation. However, the collection of such samples is often invasive, harmful, and not repeatable.
To overcome this limitation, a valid and non-invasive alternative to tissue sampling is represented by liquid biopsy from bodily fluids. The concept behind liquid biopsy is that tumours shed cellular material originating from cell death and release it into the bloodstream. It is therefore possible to analyse the blood to investigate any sort of tumour-related analyte, such as DNA, RNA, miRNAs, or microvesicles, in order to obtain information about the genotypic and phenotypic characteristics of the tumour.
The most common biological source for liquid biopsy is plasma cell-free DNA (cfDNA), obtained from blood after the removal of the cellular fraction via centrifugation. Unlike tissue samples, plasma can be easily collected at different timepoints to closely follow tumour evolution, with limited harm and risks for the patient. However, the analysis of cfDNA is challenging as its concentration is very low and cfDNA is highly degraded, with an enrichment of ~169 bp fragments. Moreover, tumour-derived cfDNA can be contaminated by DNA from healthy blood cells, and its fraction may vary from 0.01% to 60%, depending on tumour features such as volume, stage, vascularization, cell death, and proliferation.
Experimental design and results
In this research, we focused on the detection of copy number variations (CNVs) from cfDNA using a shallow whole-genome sequencing approach (sWGS) (Figure 1). sWGS is a read-count based approach able to detect CNVs from low-coverage whole genome sequencing (<1X), and it is the perfect application for nanopore technology since it is compatible with the MinION’s throughput. Also, the PCR-free nature of nanopore sequencing avoids any bias related to PCR amplification during library preparation.
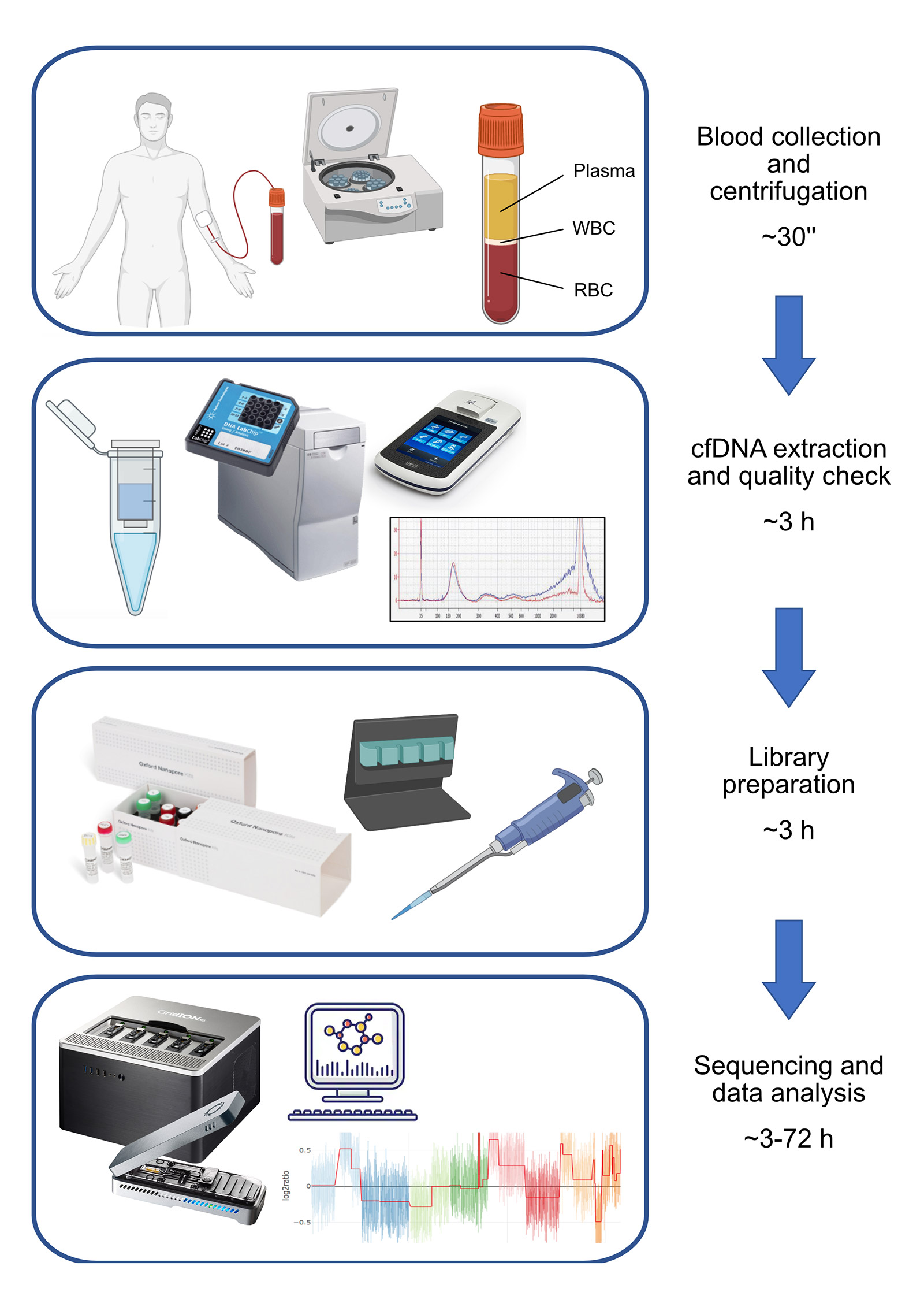
Considering the molecular characteristics of cfDNA, the choice of Oxford Nanopore as the sequencing platform may sound counter-intuitive, since it is designed to work with high quantities of long DNA fragments. It was indeed necessary to customize standard Oxford Nanopore protocols in order to adapt them to the highly degraded cfDNA; the crucial aspect consists of increasing the volume of clean-up beads to retain smaller DNA fragments. Our approach also allows multiplexing to optimise a flow cell’s sequencing power and reduce per-sample sequencing cost. However, we noticed variability in library preparation efficiency and, consequently, in per-sample throughput.
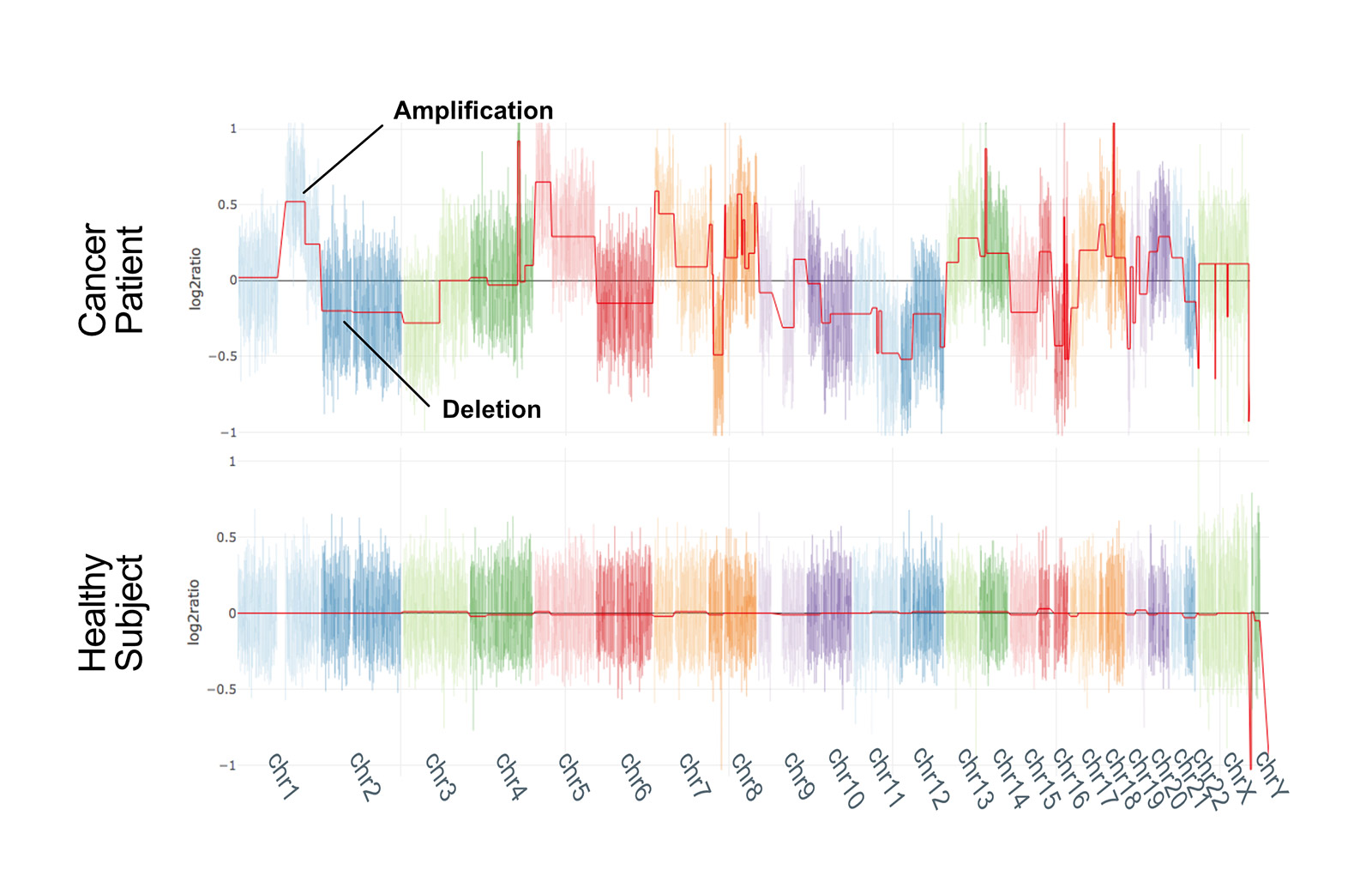
We compared nanopore technology with second generation sequencing (SGS) to validate our method: the two techniques are largely concordant and CNV profiles of samples analysed with both the approaches are highly correlated.
Our approach produces reliable results even with a limited number of reads (2 million), which means that, even though we have currently multiplexed a maximum of 5 clinical research samples, it should be feasible to multiplex up to 15 samples in a single run (~31 million reads).
Notably, 2 million reads are produced in the first 3 hours of sequencing, making this approach probably one of the fastest whole-genome CNV analysis workflows. This aspect, in combination with the possibility of performing real-time analysis, offers great future potential for the rapid analysis of clinical samples*. Notably, such performances are obtained in a singleplex run, which may not be the ideal solution in terms of per-sample costs if not dealing with urgent samples.
Why nanopore?
Sequencing-based analysis of cfDNA is currently performed with second generation sequencers, whose prices range from tens to hundreds of thousands of euros, and they are cost effective only when dealing with a large number of samples.
On the other hand, Oxford Nanopore sequencing devices (in particular the MinION) are inexpensive; reduced overall instrumentation costs make this approach accessible to most laboratories, which would otherwise be forced to outsource the sequencing, or to gain access to shared sequencers, leading often to long queues and delays. Also, Nanopore technology is extremely scalable, and only a modest number of samples is required to fill up a multiplexed run, leading to short recruitment times and, consequently, faster results.
Due to the limitations of SGS technology, sequencing-based analysis of cfDNA is currently uncommon and often limited to large research hospitals and institutions. We believe the use of nanopore sequencers would strongly contribute to the widespread analysis of cfDNA, enhancing the potential of liquid biopsy both for research and, in the future, clinical purposes*.
Future perspectives
It should be technically feasible to exploit our method to also detect single nucleotide variations (SNVs). However the relatively limited throughput of MinION and error rate are obstacles, due to the extremely low fraction of tumoral DNA in cfDNA.
We tested our approach also on Flongle Flow Cells to further reduce the per-sample cost but, unfortunately, the yield of the runs is currently insufficient.
We are confident that, with an improvement of the technology, Oxford Nanopore sequencing will overcome these obstacles in future.
*Oxford Nanopore Technologies products are currently for research use only.

Reference
- Martignano, F. et al. Nanopore sequencing from liquid biopsy: analysis of copy number variations from cell-free DNA of lung cancer patients. bioRxiv. DOI: https://doi.org/10.1101/2020.06.22.165555 (2020).






