KeyGene — Next-level crop reference genomes using plant-trained basecallers and the latest sequencing chemistry
In conjunction with this blog post, KeyGene has generated publically accessible assemblies and raw data for A. thaliana ecotype Columbia, and Tomato Heinz 1706. See here for full details
Crop reference genomes enable faster variety development
Scientists at KeyGene in the Netherlands are at the forefront of technology innovation for crop improvement. A significant focus is crop improvement through breeding for traits such as pathogen resistance, extended shelf life, and improved taste and color.
Genome sequencing has become an indispensable tool in plant breeding research, through which genetic variation of individual varieties at nucleotide precision can be revealed. Plant breeding companies use reference genomes to:
- effectively develop trait-linked markers and discover genes
- perform comparative genomics to elucidate breeding history
- dissect the genetic architecture of important traits and link them to haplotypes
- expand trait discovery approaches beyond SNP-based analysis
The insights obtained using a reference genome enable better and faster selection of important breeding trait values in the breeding process and therefore allow to bring new varieties to the market faster.
Correctness, Completeness, Contiguity and Cost, (also dubbed as the 4C’s) are of main importance with regard to generating reference genomes. High contiguity (i.e. Telomere-to-Telomere) and completeness can already be obtained by isolating High Molecular Weight (HMW) genomic DNA, in combination with (ultra) long Oxford Nanopore sequencing1. Until recently, short-read sequencing or PacBio HiFi reads were required to obtain sufficiently high consensus accuracies for plant genomes. With the introduction of the Q20+ chemistry and new nanopore types (R10.3 and R10.4) scientists have shown that near-perfect bacterial genomes can be obtained with nanopore-only data2. At KeyGene we evaluated these new pores and chemistry to improve the consensus accuracy of lettuce- and melon genome assemblies generated with nanopore-only data, and evaluated the impact of a plant-trained basecalling model on accuracy and contiguity. Finally, we evaluated the power of highly accurate duplex consensus reads, and achieved tremendous improvements of assembly quality metrics.
Experimental design and results
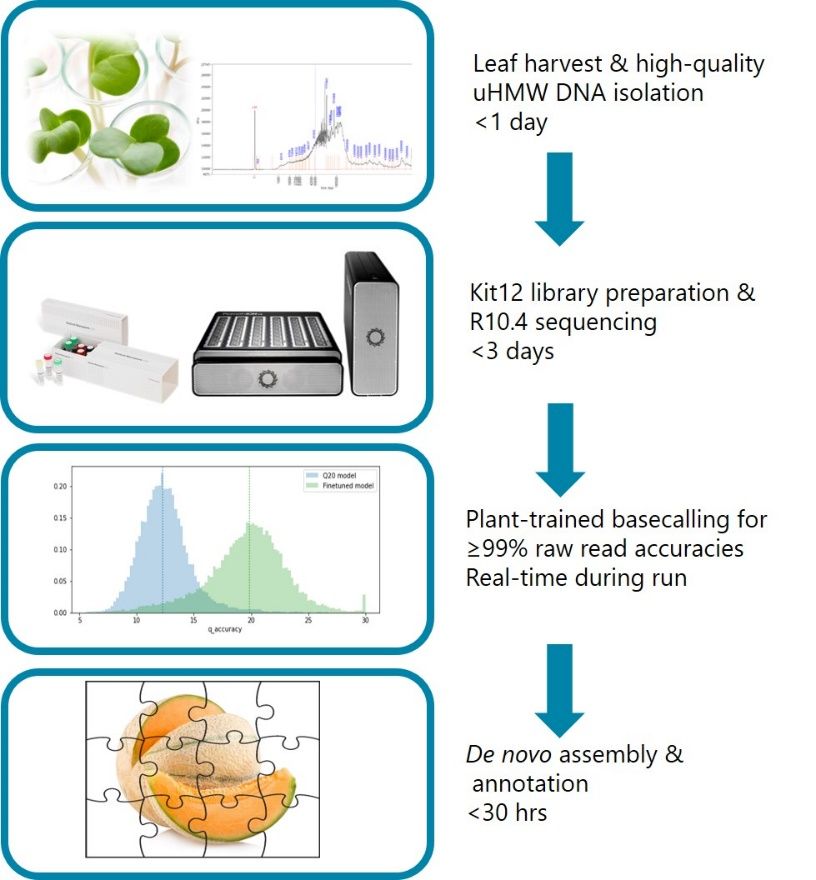
At KeyGene we have implemented a workflow for generating nanopore-only crop reference genomes within a week (Figure 1). The workflow benefits from making use of KeyGene expertise in HMW DNA extraction from a wide range of species, a plant-trained basecaller and a fast and state-of-the-art de novo assembly and annotation pipeline.
Plant-trained basecaller
Plant DNA is dramatically different from e.g. human and bacterial DNA, having a higher abundance and diversity in base modifications that negatively impact the accuracy of basecalling. To remedy this, a plant-trained basecaller for the latest Q20+ chemistry was developed in collaboration with Oxford Nanopore Technologies by augmenting the existing basecalling model with maize (B73) data. The public gold standard reference B73 RefGen_v5 was used as ground truth.
The improved model was validated by KeyGene on maize, lettuce and melon (Figure 2). As expected, most improvement in basecalling was observed with maize data, but significant basecalling improvement was also observed with sequence data from the other two, evolutionarily highly divergent, crops.
Figure 1: Workflow overview from leaf harvest to
bioinformatic analysis within a week.
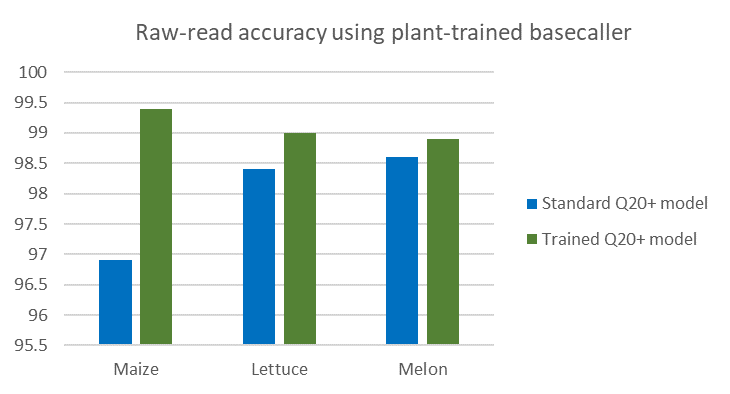
Figure 2: Median % identity alignment accuracy against golden standard reference genome using the R10.3 pore type.
Lettuce and melon reference genomes
The team at KeyGene performed whole-genome sequencing of a lettuce line using the high-yield, high-throughput PromethION platform. A total of ~75X genome coverage, with a raw read N50 of 50 Kb, was obtained using the latest Q20+ chemistry in combination with the R10.3 pore type and the plant-trained basecaller. A de novo assembly using KeyGene developed, proprietary computational tools was performed and compared to a PacBio HiFi-based assembly (Table 1).
Table 1: Assembly specifications of a lettuce line with Oxford Nanopore Technologies data compared with PacBio HiFi
| Data type | ONT R9.4.1 2018 | ONT R10.3 Q20+ | PacBio HiFi |
| Analysis method | Minimap Miniasm | KeyGene STL | Hifiasm v0.16.1 |
| Total assembly size (Gb) | 2.68 | 2.59 | 2.63 |
| N50 contig size (Mb) | 6.38 | 50.31 | 11.97 |
| N50 index | 127 | 16 | 62 |
| Max contig size (Mb) | 26 | 132.5 | 49.1 |
| QV score (Mercury) | 28 | 47 | 52 |
The KeyGene® STL assembler generated a reference genome that is 8x more contiguous than the reference genome that KeyGene generated using the R9.4.1 pore type in 20183 from the same line and 4x more contiguous compared to a PacBio HiFi assembly. The number of contigs dramatically decreased from 153,952 as generated by the public reference4, down to 159 generated by our current nanopore-only assembly. With regard to consensus accuracy, the nanopore-only assembly is on par with the PacBio HiFi-based assembly and was obtained within 30 hours after data collection.
Cantaloupe melons (Cucumis melo var. cantalupo) with their netted green and beige skin and deliciously sweet orange flesh are one of the most popular types for consumption. We used the R10.4 pore type with Q20+ chemistry to generate a nanopore simplex dataset of 169 Gb with a raw read N50 of 31 Kb. From this dataset we could extract 16 Gb of duplex consensus data with raw modal read accuracies of 99.9%. De novo assemblies of simplex- and duplex nanopore reads were generated using Flye5 as well as Keygene’s STL assembler. The results were compared to a HiFi-based assembly generated by the Hifiasm assembler6 (Table 2).
Table 2: Assembly specifications of Cantaloupe melon from Oxford Nanopore simplex (20X coverage) and duplex (35X coverage) compared to a HiFi-based (20X coverage) assembly
| Data type | ONT R10.4 Q20+ | ONT R10.4 Q20+ | ONT R10.4 duplex | PacBio HiFi |
| Analysis method | Flye 2.9 --nano-hq | KeyGene STL | Flye 2.9 --nano-hq | Hifiasm v0.16.1 |
| Assembly size (Mb) | 365 | 367 | 371 | 392 |
| N50 contig size (Mb) | 13.6 | 13.6 | 13.4 | 11.1 |
| N50 index | 8 | 9 | 9 | 10 |
| Max contig size (Mb) | 27.7 | 26.9 | 26.9 | 39.6 |
| QV score | 49 | 46 | 52 | 49 |
The data shows that all nanopore-only assemblies have an improved contiguity (N50 index ≤ 9) when compared to the HiFi-based assembly at similar genome coverage. With regard to the consensus accuracy a similar or even better accuracy could be obtained for the nanopore-only Flye assemblies. These results showcase the improved strength of nanopore data for generating highly accurate and contiguous plant reference genomes, without the help of additional sequence data.
Data releases
In conjunction with the work shown above, we at KeyGene have applied the latest chemistry and basecalling improvements to two additional plant species: 1) Arabidopsis thaliana ecotype Columbia and 2) Tomato Heinz 1706; see Table 3 for a summary of the dataset features. For full details on these datasets, including download locations and experimental approaches, please see the article here.
Table 3: Dataset properties for Arabidopsis thaliana ecotype Columbia and Tomato Heinz 1706, as generated and released by KeyGene
| Dataset | Flow cell ID | Pass data yield (Gb) | N50 read length | Median % identity |
| A. thaliana Col-0 | FAR28243 | 14.1 | 36.6 | 96.9 |
| Tomato Heinz 1706 | PAI84774 | 55.6 | 48.4 | 98.1 |
| Tomato Heinz 1706 | PAI85364 | 52.2 | 49.3 | 98.1 |
| *Calculated median percentage identity using Nanoplot against latest public reference genome for respective species. | ||||
Future perspectives
Here, we show for the first time that using a plant-trained basecalling model, nanopore-only reference crop genomes can be obtained with outstanding contiguity and accuracy, reducing the requirements for multiple technologies to generate reference-quality genomes. Ongoing developments are focused on increasing the percentage and length of duplex reads as well as further enhancing the speed of basecalling. Plant trained basecallers will further improve with the addition of more native plant context in the near future. From a data analysis perspective KeyGene is focusing on telomere-to-telomere assemblies of crop genomes using only data from the Oxford Nanopore Technologies platform, and enabling the full phasing of heterozygous and polyploid genomes7,8.
We are extremely excited about the future and and truly believe that this spectacular increase in assembly quality metrics will pave the way to democratize genome sequencing in crop improvement by using the rapid, low-cost and easy-to-use nanopore platforms.
More information
KeyGene is a Commercial Service Provider for nanopore sequencing and is a paying member of the Oxford Nanopore Service Provider Programme. To read more about their service offerings see here.
- Purchase R10.4 flow cells through the nanopore store
- Visit the KeyGene website
- Read more about the KeyGene dataset release
- Closing the gap in plant genomes white paper https://nanoporetech.com/resource-centre/closing-gap-plant-genomes-white-paper (2021).
- Oxford Nanopore R10.4 long-read sequencing enables near-perfect bacterial genomes from pure cultures and metagenomes without short-read or reference polishing. bioRxiv. https://www.biorxiv.org/content/10.1101/2021.10.27.466057v2 (2021).
- The tip of the iceberg — Sequencing the lettuce genome https://nanoporetech.com/resource-centre/tip-iceberg-sequencing-lettuce-genome
- Sebastian Reyes-Chin-Wo, Zhiwen Wang, Xinhua Yang, et al. Genome assembly with in vitro proximity ligation data and whole-genome triplication in lettuce. Nature Communications. DOI: https://doi.org/10.1038/ncomms14953 (2017).
- Mikhail Kolmogorov, Jeffrey Yuan, Yu Lin and Pavel Pevzner, "Assembly of Long Error-Prone Reads Using Repeat Graphs". Nature Biotechnology. DOI:10.1038/s41587-019-0072-8 (2019).
- Cheng, H., Concepcion, G.T., Feng, X., Zhang, H., Li H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods: 18:170-175. https://doi.org/10.1038/s41592-020-01056-5 (2021).
- Phased Genome Sequencing “Opportunities for more effective crop breeding”; Prophyta annual (2020).
- Thomas W. Wöhner, Ofere F. Emeriewen, Alexander H.J. Wittenberg et al. The draft chromosome-level genome assembly of tetraploid ground cherry (Prunus fruticosa Pall.) from long reads. Genomics. https://doi.org/10.1016/j.ygeno.2021.11.002 (2021).






