Oxford Nanopore sequencing accuracy

For many years we have iterated our technology to improve performance. We continue to do so through updates to analytical methods and chemistries. We continue to improve the nanopore sensing system, through updates to analytical methods and new chemistries. This page guides you on what to expect from the nanopore sequencing system, and which tools to choose to achieve these results.
What is sequencing accuracy?
Accuracy is a generic term that might refer to different aspects of DNA and RNA sequencing performance. Typically, it refers to the accuracy at a single read level or at the consensus level, combining the information from multiple reads of a DNA/RNA region into a single high-quality sequence. Depending on the application, other relevant factors to consider are the proportion of the genome covered and the ability to detect epigenetic modifications. Usually, genomic research focuses either on resequencing and mapping to a reference genome or reconstructing unknown genomes through de novo assembly, assembly. For mapping-based projects, changes compared with the reference sequences are used for inference, hence variant calling becomes the main focus. For de novo quality is estimated by the accuracy of the reconstructed sequence and other metrics such as N50.
Variant calling accuracy
Variant calling identifies differences from a reference sequence and is crucial in understanding how genotypes drive phenotypes. Oxford Nanopore technology can sequence any length of DNA and RNA molecule, offering unprecedented resolution of complex structural variants and efficient haplotype phasing of variants.
Measuring the accuracy of variant calling is critical to ensure that the genetic variants identified are biological differences and not artefacts. Accuracy is commonly measured with the so called F1 score, the harmonic mean of precision (proportion of called variants that are actually variants) and sensitivity or recall (proportion of all variants that are correctly called). This metric is especially useful when you want to balance the trade-off between identifying as many variants as possible (high sensitivity) and ensuring the variants identified are truly variants (high precision).
Our SNP calls produced with [nanopore sequencing] … were comparable to state-of-the-art short-read-based methods
Kolmogorov et al. Nat. Methods (2023)
[With nanopore Q20 chemistry] It is now realistic to use long read sequencers to systematically analyze a wider range of cancerous mutations
Sakamoto et al. Nat. Commun. (2022)
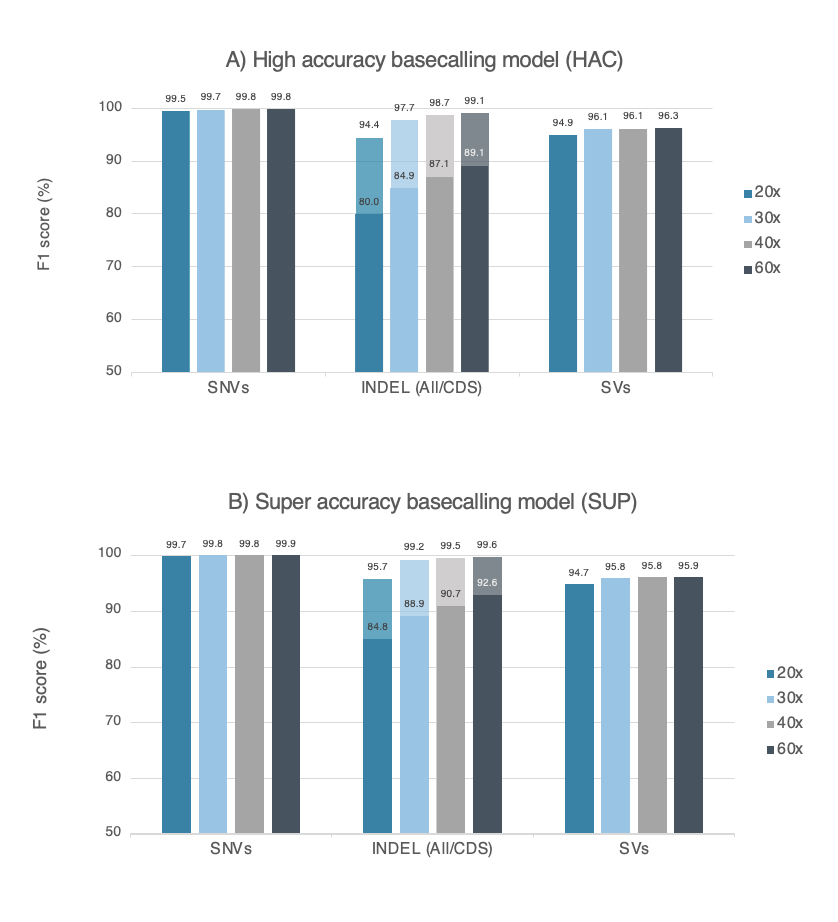
Figure 1. Accuracy data obtained from a dataset of 29kb N50 prepared with the Ligation Sequencing Kit V14 (with enzyme E8.2.1) and PromethION R10.4.1 Flow Cell. Accuracy is measured as F1 score for variant calling, using nanopore sequencing data for the human genome (HG002 cell lines) at several read depths. Variant calling was performed with wf-human-variation workflow version v2.2.6, and variants were compared against the Genome In A Bottle consortium’s HG002 truth-set (SNVs and Indels v4.2.1, SVs v0.6) . SNVs and indels (< 50bp) are represented with dark colours, while indels (< 50bp) in coding regions (CDS) are displayed with lighter colours. F1 score for Dorado basecalling models of A) high accuracy (HAC, v5.2.0) and B) super accuracy (SUP, v5.2.0).

Base modification accuracy
The four DNA bases (A, C, G, T) and RNA bases (A, C, G, U) can undergo biological modifications like methylation, impacting gene expression and contributing to diseases such as cancer. Oxford Nanopore’s technology allows for direct, real-time sequencing and detection of these modifications for both DNA and RNA (e.g. 5mC, 5hmC, 6mA, 4mC for DNA, m6A, and pseudoU for RNA) without additional experiments or preparation, unlike legacy methods, such as bisulphite sequencing, that have several limitations.
Read more about direct DNA and RNA base modifications detection
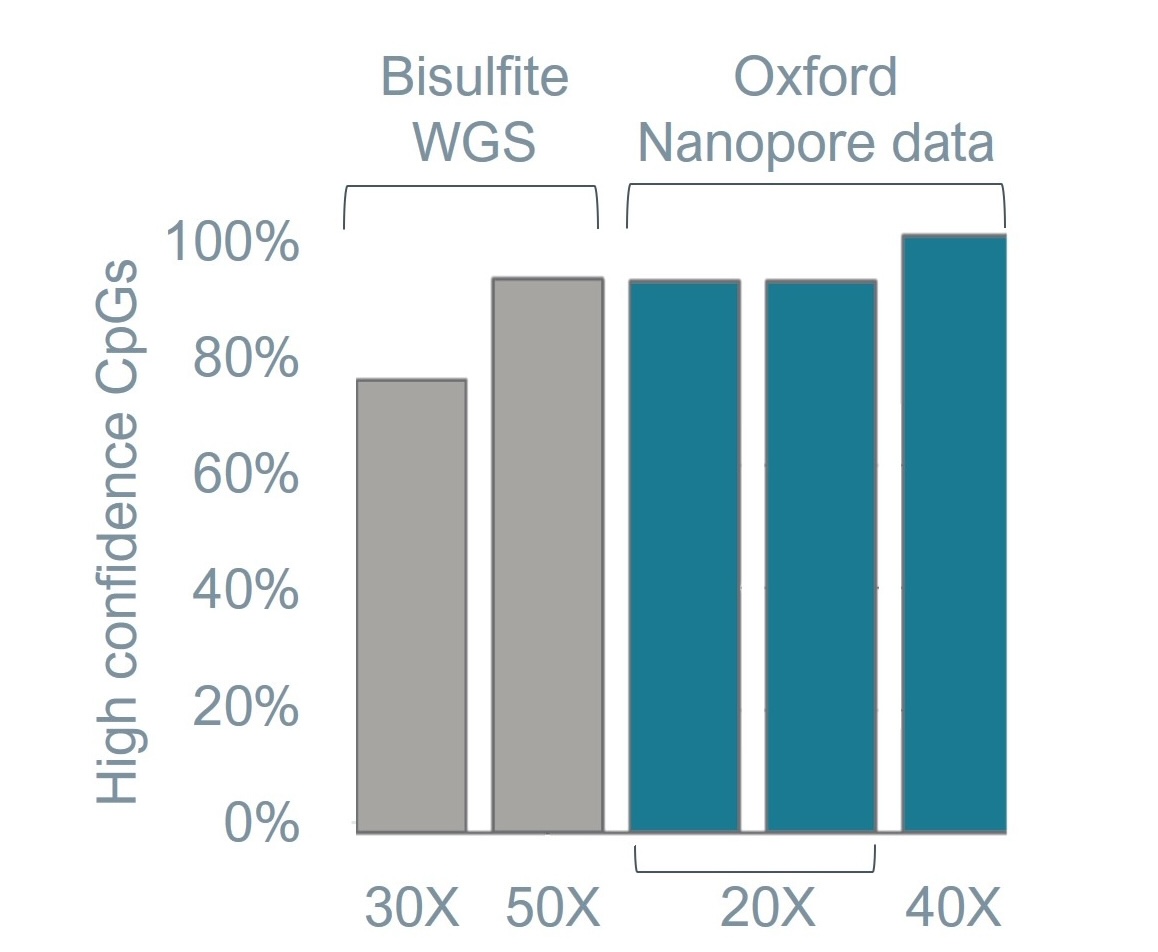
Figure 2. Bisulfite sequencing data. Basecalling of 5mC on synthetic strands with known composition is extremely accurate with precision, recall, and F1 score all above 99%. Oxford Nanopore data for the human sample HG002 shows much higher confidence CpGs (>90%) at a much lower depth than bisufite whole-genome sequencing (WGS). All data reported in this figure was generated with Ligation Sequencing Kit V14 and PromethION R10.4.1 Flow Cells using SUP basecalling models.
Our methylation calls [with nanopore sequencing] were highly concordant with the standard bisulfite sequencing, but in addition had haplotype-specific resolution
Kolmogorov et al. Nat. Methods (2023)
| Molecule | Modification | Molecular context | Raw read accuracy (SUP) |
|---|---|---|---|
| DNA | 5mC | CpG | 99.5% |
| 5mC | All | 99.4% | |
| 5mC/5hmC | CpG | 99.2% | |
| 5mC/5hmC | All | 98.7% | |
| 6mA | All | 99.7% | |
| 4mC/5mC | All | 97.6% | |
| RNA | m6A | DRACH | 99.7% |
| m6A | All | 98.7% | |
| pseU | All | 97.6% | |
| m5C | All | 97.9% | |
| Inosine | All | 98.8% | |
| 2’OMe-A | All | 99.2% | |
| 2’OMe-C | All | 98.7% | |
| 2’OMe-G | All | 98.2% | |
| 2’OMe-U | All | 96.7% |
Table 1. Currently supported models for DNA and RNA modification basecalling available in Dorado standalone in GitHub. Accuracy values were generated on a synthetic truth-set using v5.2 SUP (for DNA) and v5.2 SUP (for RNA) basecalling models. All modification models, except RNA 2’OMe are currently available in the latest MinKNOW version. 2'OMe models will be integrated in later MinKNOW versions.
Assembly accuracy
Assembly accuracy refers to the degree to which a reconstructed sequence of DNA or RNA matches the true biological sequence from which it was derived. This involves building a consensus sequence from multiple DNA/RNA reads, enhancing accuracy and creating a reliable sequence for further analysis.
Find out more about assembly & whole-genome sequencing.
“Even for lower (<30×) coverage ONT-only datasets, genome-wide consensus accuracy exceeds both the standard recommended by the Vertebrate Genomes Project (QV40) and the genome-wide consensus accuracy we previously reported for a D. melanogaster R9.4.1 and Illumina hybrid assembly.”
Kim et al., PLOS Biology (2024)
Oxford Nanopore sequencing achieves:
| Flow cell | Library preparation kit | Assembly accuracy | Sequencing & basecalling parameters | Analysis tools | Sample |
|---|---|---|---|---|---|
| PromethION R10.4.1 | Ultra-long Sequencing Kit V14 (ULK), Ligation Sequencing Kit V14 (for Pore-C), and Assembly Polishing Kit (APK) V141 | Telomere-to-telomere (T2T): Q512, 30 full chromosome haplotype-resolved, N50 >144 Mb | Simplex SUP (ULK and APK3) simplex HAC (Pore-C) | Read correction with Dorado correct4, assembly with Verkko, phasing with Gfase, polishing with Medaka | Human HG002 |
| MinION R10.4.1 | Ligation Sequencing Kit V14 | Q50 at 10–20x | Simplex SUP | Assembly with Flye | Zymo mock community (bacterial) |
1APK available as part of the T2T bundle, register your interest here.
2Generated by combining approx. 45x ultra-long (2 PromethION Flow Cells), 35x Pore-C (1 PromethION Flow Cell), and 35x assembly polishing data (1 PromethION Flow Cell). View full dataset.
3Specific basecalling model for APK.
4Based on the HERRO tool (Stanojević et al. 2024)
Covering all of the genome
To create an accurate picture of the genome, it is important for a sequencing technology to reach all parts of it, even the parts which are difficult to map. Genomes are littered with repetitive and low-complexity regions, which are difficult to sequence and align using legacy technologies. For example, it is estimated that short-read technology reaches only 92% of the human genome, leaving 8% that contains many disease-relevant genes excluded from the dataset.
Oxford Nanopore technology has been shown to reduce these ‘dark’ areas of the genome by 81%, shedding light on parts of the genome not sequenced by any other technology (Ebbert et al., 2019), and giving a more complete picture. The extensive genome mapping capabilities of nanopore data manage to achieve 99.49% genome coverage (Uddin et al., 2024). Ultra-long nanopore sequencing reads were central to completing the human genome, resolving repetitive regions that were unattainable with other technologies (Nurk et al., 2022).
[Nanopore] sequencing allowed us to unravel the only two unsolved cases … for which different genetic testing approaches had been sequentially performed for > 10 years unsuccessfully
Damián et al. Hum. Genomics (2023)
Raw read and single molecule accuracy
Oxford Nanopore sequencing uses direct electronic analysis of native DNA and RNA molecules to generate raw reads, eliminating PCR bias. Basecalling algorithms based on machine learning have been improving with time, providing more and more accurate reads. Raw read accuracy refers to the accuracy achieved when reading a single DNA or RNA strand once. Most applications focus on variant calling, consensus accuracy, or other metrics, where the information from several reads is combined. These can be improved by increased raw read accuracy but can also be enhanced in other ways (e.g. increased genome coverage).
Nanopore sequencing achieves:
Oxford Nanopore sequencing hardware and chemistry have seen major upgrades in the shift to R10.4.1 and are now able to read DNA fragments at >99% single-read accuracy (the “Q20 chemistry”).
Kim et al., PLOS Biology (2024)
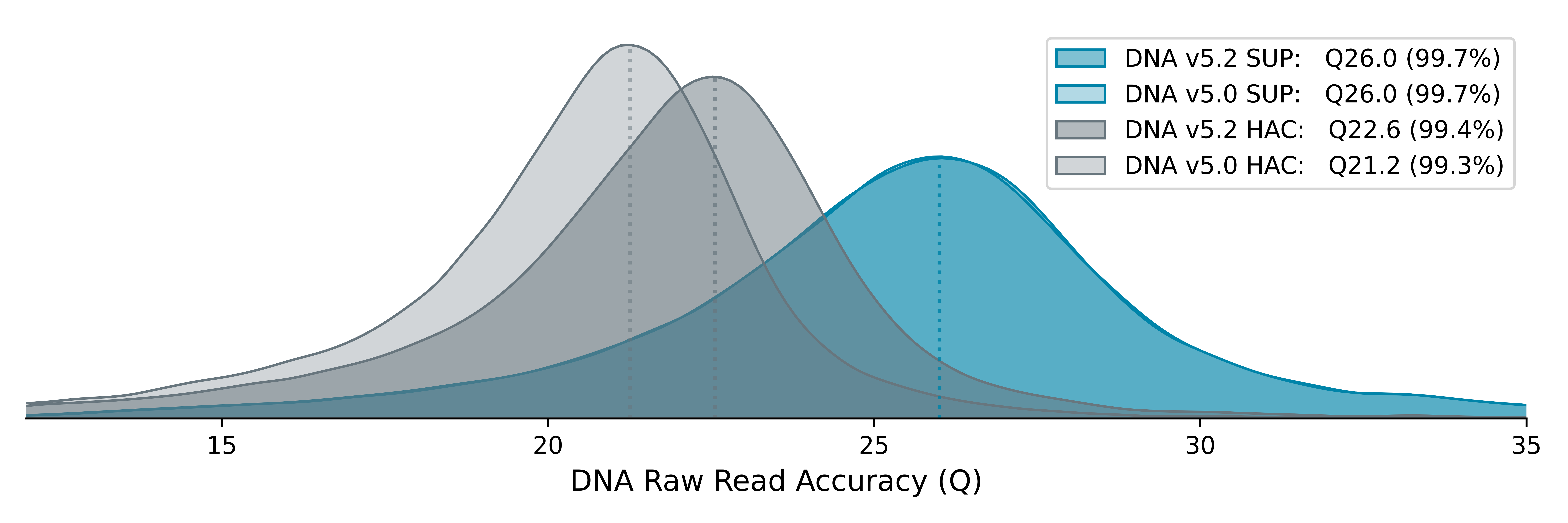
Figure 3. DNA raw read accuracy modal data obtained with Ligation Sequencing Kit V14 (with enzyme E8.2.1) and PromethION R10.4.1 Flow Cells, using nanopore sequencing data for the human genome (HG002 cell lines). Both HAC and SUP models are featured for version 5.0 and 5.2 (integrated in MinKNOW).
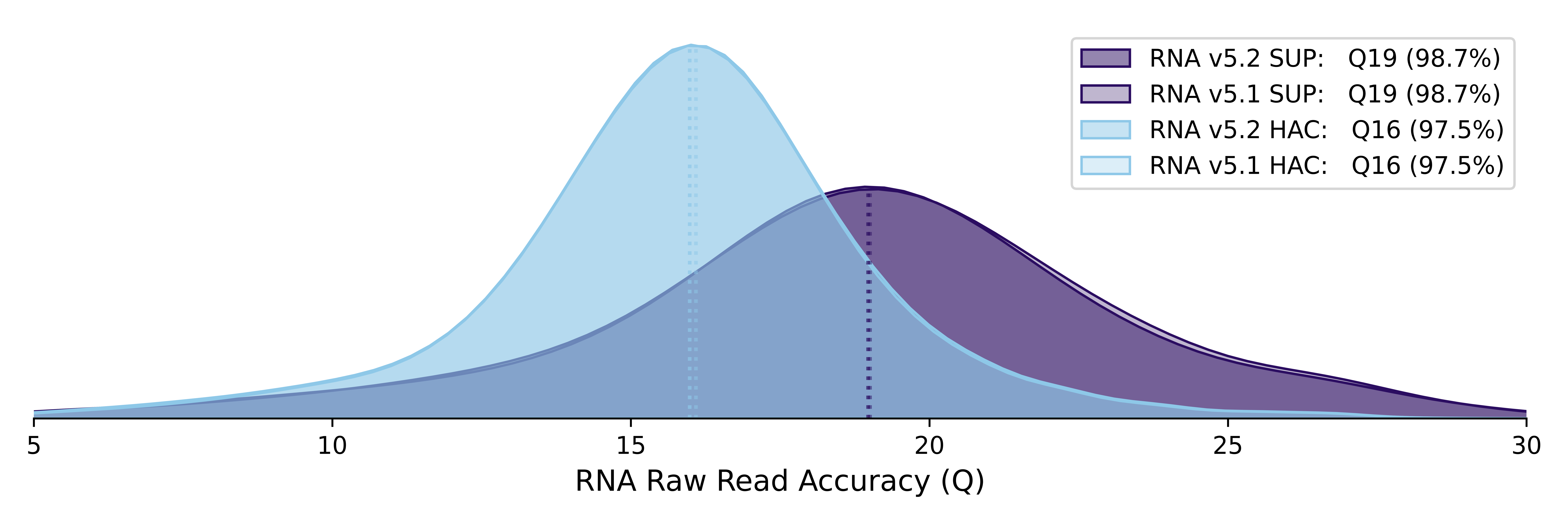
Figure 4. RNA raw read accuracy modal data obtained with Direct RNA Sequencing Kit (RNA004) and PromethION RNA Flow Cells, using nanopore sequencing data for the UHRR total RNA sample input. Both HAC and SUP models are featured for version 5.1 (currently integrated in MinKNOW) and 5.2 (currently available in Dorado standalone in GitHub).
Tuning accuracy for your experimental need
Optimise accuracy according to your requirements by selecting the most suitable basecalling model.
Fast basecalling: fastest, least computationally intense. Compatible with real-time basecalling on all nanopore devices with compute. Recommended for quick, real-time insights on sequencing data when compute resources are limited.
High accuracy basecalling (HAC): highly accurate, intermediate speed and computational requirement. Compatible with real-time basecalling on GridION and PromethION devices with compute. Recommended for high-throughput projects focusing on variant analysis.
Super accuracy basecalling (SUP): the most accurate and computationally intense. Recommended for de novo assembly projects and low-frequency variant analysis (e.g. somatic variation and single-cell applications).
Duplex basecalling: is recommended for hemi-methylation investigation, enabling the methylation signature of each DNA strand to be distinguished. This is available via Dorado standalone in GitHub.
Oxford Nanopore raw reads now achieve 99.75% (Q26) accuracy with the latest Dorado basecalling models (v5) available in GitHub.
Available datasets
Oxford Nanopore Technologies provides open access to a range of nanopore sequencing datasets through its initiative hosted on Amazon Web Services (AWS), called ‘ont-open-data’. This initiative allows researchers worldwide to explore and utilise extensive sequencing data to enhance their genomic studies. For example, the dataset for the human genome sample GM24385 (HG002) is one of the available resources, which has been utilised in numerous research applications, reflecting Oxford Nanopore's commitment to supporting the scientific community by providing freely accessible, high-quality data.
Get in touch
Subscribe
Talk to us
If you have any questions about our products or services, chat directly with a member of our sales team.






