On the quest to assemble the giant lungfish genome
Our closest relative
Considered a “living fossil”, the Australian lungfish (Neoceratodus forsteri) was discovered just a little more than 150 years ago and was at first misclassified as an amphibian due to its peculiar body. These fishes are equipped with fleshy lobed fins which they use to crawl on the bottom of rivers and they can also breathe air through their rudimentary lungs. We now know that these fascinating fishes are the closest living relatives of all tetrapods, including humans, making them of great interest to evolutionary biologists. Their unique morphology, physiology and biochemistry can help us understand the mechanisms that enabled aquatic vertebrates to “conquer” the land back in the Devonian period (about 400 million years ago).
The quest for the lungfish genome
In the last few decades, researchers have used different molecular techniques to access and leverage the information contained in parts of the lungfish DNA (mainly coding regions). To gain a better understanding of the molecular mechanisms underpinning the innovations associated with terrestrialization, a more complete understanding of the lungfish’s genome was needed. However, sequencing the lungfish genome posed an extreme challenge due to its incredible size, estimated to be more than 16 times larger than the human genome according to flow cytometry- and photometry-based measurements (Pedersen 1971; Rock, et al. 1996). It was deemed impossible…until now. The tremendous progress in long-read sequencing technologies has recently provided us with a powerful toolbox to embark on such a challenging project – the sequencing of the largest vertebrate genome. After securing funding and establishing a team of researchers with skills in genome assembly and comparative genomics, we chose Oxford Nanopore for generating sequence data. Our choice was driven by the fact that Nanopore ultra-long reads have the potential to resolve a large fraction of the repetitive elements presumed to constitute the majority of this huge genome. Long repetitive regions seem to be a common signature of large eukaryotic genomes, as for example in the Mexican salamander axolotl (Nowoshilow, et al. 2018).

What the lungfish genome tells us about conquering the land
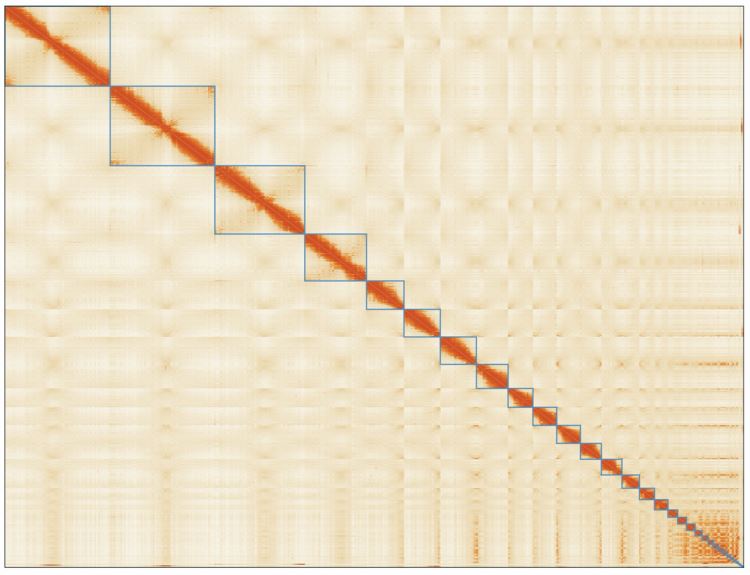
We sequenced 1,200 billion base pairs of the Australian lungfish DNA on the PromethION platform, which yielded an assembly of 62 thousand contigs (N50 of 1.86 Mb). These were then polished and scaffolded using short-reads obtained with chromosome conformation capture techniques (Hi-C), enabling a chromosome-level assembly (N50 1.7 Gb). This lungfish genome is now the largest animal genome sequenced to date. It is nearly 14 times larger than the human genome, and includes 43 billion base pairs distributed in 27 large scaffolds which correspond to the chromosome sizes established using cytogenetics in 1996 (Rock, et al. 1996). By using comparative genomics, we found that this fish possesses genetic pre-adaptations for terrestrial conditions, such as genetic elements that are crucial for the evolution of limbs from fins, and for breathing air and olfaction. Given the high-quality of our assembly, we were also able to characterize the repetitive elements constituting approximately 90% of the total genetic material. Notably, we identified a tetrapod-like signature in these elements, as their families and relative abundances more closely resemble those of land vertebrates than fishes.
Overall, we demonstrated how Nanopore sequencing provides a suitable and reliable tool for sequencing giant genomes.
| Estimated Chromosome Size (Gb) | Scaffold Size (Gb) |
|---|---|
| 5.40 | 4.92 |
| 5.40 | 4.89 |
| 5.20 | 4.21 |
| 2.50 | 2.78 |
| 2.20 | 1.75 |
| 1.90 | 1.69 |
| 1.90 | 1.69 |
| 1.90 | 1.48 |
| 1.60 | 1.16 |
| 1.60 | 1.11 |
| 1.60 | 1.10 |
| 1.30 | 0.98 |
| 1.30 | 0.96 |
| 1.20 | 0.82 |
| 1.10 | 0.70 |
| 0.80 | 0.63 |
| 0.70 | 0.45 |
| 0.60 | 0.41 |
| 0.60 | 0.40 |
| 0.60 | 0.33 |
| 0.50 | 0.27 |
| 0.50 | 0.24 |
| 0.40 | 0.24 |
| 0.40 | 0.22 |
| 0.40 | 0.19 |
| 0.20 | 0.15 |
| 0.20 | 0.15 |
- Nowoshilow S, Schloissnig S, Fei JF, Dahl A, Pang AWC, Pippel M, Winkler S, Hastie AR, Young G, Roscito JG, et al. 2018. The axolotl genome and the evolution of key tissue formation regulators. Nature 554:50-55.
- Zimin A, Stevens KA, Crepeau MW, Holtz-Morris A, Koriabine M, Marçais G, Puiu D, Roberts M, Wegrzyn JL, de Jong PJ, Neale DB. Sequencing and assembly of the 22-Gb loblolly pine genome. Genetics. 2014 Mar 1;196(3):875-90.
- Kolmogorov M, Yuan J, Lin Y, Pevzner PA. Assembly of long, error-prone reads using repeat graphs. Nature biotechnology. 2019 May;37(5):540.
- Miller JR, Delcher AL, Koren S, Venter E, Walenz BP, Brownley A, Johnson J, Li K, Mobarry C, Sutton G. Aggressive assembly of pyrosequencing reads with mates. Bioinformatics. 2008 Oct 24;24(24):2818-24.
- Zimin AV, Salzberg SL. The genome polishing tool POLCA makes fast and accurate corrections in genome assemblies. bioRxiv. 2019 Jan 1.






